








全ゲノム解析で明らかになる日本人の遺伝的起源と特徴
-ネアンデルタール人・デニソワ人の遺伝子混入と自然選択-
2024年4月18日 理化学研究所
理化学研究所(理研)生命医科学研究センター ゲノム解析応用研究チームの寺尾 知可史 チームリーダー(静岡県立総合病院 臨床研究部 免疫研究部長、静岡県立大学 薬学部ゲノム病態解析講座 特任教授)、劉 暁渓 上級研究員(研究当時:ゲノム解析応用研究チーム 研究員; 静岡県立総合病院 臨床研究部 研究員)、東京大学医科学研究所附属ヒトゲノム解析センター シークエンス技術開発分野の松田 浩一 特任教授らの共同研究グループは、大規模な日本人の全ゲノムシークエンス(WGS)[1]情報を分析し、日本人集団の遺伝的構造、ネアンデルタール人[2]およびデニソワ人[3]由来のDNAと病気の関連性、そしてゲノムの自然選択が影響を及ぼしている領域を複数発見しました。
本研究成果は、日本人集団の遺伝的特徴や起源の理解、さらには個別化医療[4]や創薬研究への貢献が期待されます。
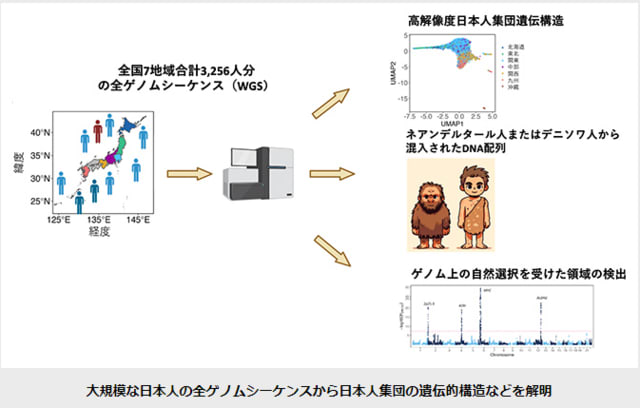
今回、共同研究グループは、バイオバンク・ジャパン(BBJ)[5]が提供した3,256人分の日本人の全ゲノム情報を分析しました。この研究を通じて、日本人の祖先に関わる三つの源流(縄文系祖先、関西系祖先、東北系祖先)の起源を明らかにしました。
また、現生人類(ホモ・サピエンス)の最も近縁とされる古代型人類ネアンデルタール人やデニソワ人から受け継いだ遺伝子領域を特定しました。一部の日本人が持つNKX6-1遺伝子領域は、2型糖尿病リスクと関連しており、この領域がデニソワ人由来であることが明らかになりました。さらに、日本人の遺伝子における自然選択が作用している領域も同定しました。
背景
全ゲノムシークエンス(WGS)データの分析を通じて、人間の遺伝的多様性や自然選択のプロセスを理解する上での新たな扉が開かれました。また、個別化医療や創薬研究におけるゲノムワイド関連解析(GWAS)の解析能力を格段に向上させ、病気に関わるバリアントの同定や治療標的の特定へとつながっています。
しかし、日本人集団を対象としたWGS研究は、限定的な規模にとどまっていました。このギャップを埋めるため、共同研究グループは全国7地域から集めた3,256人分のゲノム情報を解析し、日本人特有の遺伝的特徴を明らかにすることを企図しました。
研究手法と成果

A)サンプルが集められた日本の七つの地域。B)コモンバリアントを基にしたPCA分析を行い、参加者を集めた地域ごとに色分けした。C)レアバリアントに基づくPCA-UMAP分析を示す。B)、C)では、一つ一つの点が個人を表しており、点と点との距離は遺伝学的な違いを反映している。近い点ほど遺伝学的には近縁である。この二つの図では、出身地を元にした遺伝背景の違いにより、色分けして表示している。
D)ADMIXTURE分析を行い、三つの集団(K=3)に分けた。沖縄以外の地域からはランダムに100人を選び、沖縄からは全員(28人)をそれぞれ分析した。K1は沖縄、K2とK3はそれぞれ東北と関西で最も高い値を示す。
バイオバンク・ジャパン(BBJ)により、全国7地域(北海道、東北、関東、中部、関西、九州、沖縄)の医療機関に登録された合計3,256人分の全ゲノムシーケンス(WGS)を行って、「Japanese Encyclopedia of Whole/Exome Sequencing Library(JEWEL)」というデータセットが作成されました(図1A)。この最終データセットは、45,586,919個のSNV[6]と9,113,420個のindel[7]を含み、そのうち15,410,953個(32.7%)がJEWELで新たに観察されたバリアント[8]です。これは日本における最も包括的な全ゲノムシーケンスデータの一つです。日本人の集団構造を理解するために、まずコモンバリアント[8]に基づいて従来の主成分分析(PCA)[9]を行いました。以前の研究と同様に、この分析から沖縄と本土クラスターの二重構造が再現されました(図1B)。レアバリアント[8]が人口構造を明らかにする上でより多くの情報を提供する可能性があると仮定し、1,835,116個のレアバリアントを使用した次元削減(PCA-UMAP)分析[10]を行いました。この分析により、日本人口の前例のない遺伝構造が明らかになりました(図1C)。この構造は、コモンバリアントに基づいてPCAから得られたパターンを再現するだけでなく、いくつかの顕著な特徴を示しています。
1)本土の領域間のより明確な分離と、本土クラスターから沖縄クラスターのより明確な区別が観察されます。
2)東北の人々が細長いエリアにクラスタリングされます。
さらに、WGSのデータADMIXTURE分析[11]による解析結果は、日本人口は三つの祖先(以下、K1、K2、K3)の混合によって最もよくモデル化できることを示唆しています。K1、K2、K3はそれぞれ沖縄、東北、関西で最も高まります(図1D)。K1(沖縄)成分は、南(沖縄に隣接する地域)を除く本土のサブグループで比較的安定した割合の約12%を維持し、南ではより高い割合の22%を示しました。K2(東北)とK3(関西)成分は西から東北へ徐々に変化していくことが分かります(図1D)。
以前の研究により、日本人が縄文人および東アジア(EA、主に漢民族)の祖先を持つことが示唆されています。最近の古代ゲノムの分析からは、北東アジア(NEA)の祖先の影響も指摘されています。この背景のもと、共同研究グループは、縄文、EA、NEAの現代および古代の遺伝データを今回のデータと共に分析しました。
縄文の祖先比率については、沖縄が最も高い比率(28.5%)を持ち、次いで東北(18.9%)である一方、関西が最も低い(13.4%)と推定されました。これは、縄文人と沖縄の人々の間に高い遺伝的親和性があることを示す以前の研究と一致しています。また、関西地方は漢民族と遺伝的親和性が高いことが明らかになりました。
さらに、共同研究グループは中国、韓国、日本から報告された古代人ゲノムデータを使って、東北と関西の間の遺伝的親和性の違いを評価しました。その結果、関西人と黄河(YR)またはその上流地域の中新石器時代および後新石器時代古代中国集団との間に顕著に密接な関係があることが見受けられました。対照的に、東北地方の個体は、縄文人との遺伝的親和性が顕著に高く、また沖縄の宮古島の古代日本人ゲノム(高い縄文比率を持つ)や韓国三国時代(4~5世紀)の古代韓国人とも高い遺伝的親和性を持つことが示されました。
共同研究グループは、絶滅したネアンデルタール人やデニソワ人から引き継がれた可能性のある遺伝子配列を検出するために、最新の確率的手法IBDmix[12]を使用しました。JEWELの個々のデータ分析により、ネアンデルタール人由来の約49Mb(メガ・ベース:DNAの長さの単位で1Mbは100万塩基対)とデニソワ人由来の約1.47Mbの遺伝子配列が検出されました。合計で、ネアンデルタール人から引き継がれた可能性が高い3,079セグメント(ゲノム領域)とデニソワ人から引き継がれた可能性の高い210セグメントが特定され、それぞれ772Mbと31.46Mbのゲノムをカバーしています(図2)。

各染色体にわたり混入された配列の分布を示す密度プロット。上部のトラック(青で示される)はネアンデルタール人から直接受け継がれたと考えられる配列を表し、下部のトラック(赤で示される)はデニソワ人からの配列を示す。
BBJからGWASの結果に基づいて、引き継がれた配列が106の表現型に与える影響を調査しました。デニソワ人由来2個とネアンデルタール人由来42個を含む44の領域と49の表現型とが関連付けられました。これらのうち43の関連は、以前の研究では報告されていません。特に、POLR3Eのデニソワ人由来セグメントは身長と、NKX6-1のセグメントは2型糖尿病(T2D)と関連していました(図3)。

NKX6-1遺伝子の領域におけるデニソワ人から直接受け継がれたと考えられる変異は、日本人集団における2型糖尿病(T2D)と関連している。横軸にヒトゲノム染色体上の位置、縦軸に各変異のT2Dとの関連の強さを示した。三角形はデニソワ人から受け継がれた変異を指し、灰色の点はそれ以外の変異を示す。
さらに、ネアンデルタール人由来のセグメントはT2D、冠状動脈疾患(CAD)、安定狭心症(SAP)、アトピー性皮膚炎(AD)、グレーブス病(GD)、前立腺がん(PrCa)、関節リウマチ(RA)など七つの病気と関連する11の領域が観察されました。これらの発見は、特に東アジア人において顕著な集団特異性を示しており、日本人におけるアリル頻度[13]の中央値が欧州人と比較して21.5倍であることが分かりました。
共同研究グループは、日本人口において選択された可能性があるゲノム領域を特定するために、全ゲノムスキャンを、iHS[14]とFastSMC[15]という二つの方法で実施しました。iHS法により、共同研究グループは全ゲノム(MHC、ADHクラスター、およびALDH2を含む)の有意水準で正の選択下にある三つの領域を特定しました(図4A)。
さらに、関西、関東、東北、九州、沖縄の五つの代表的な地域を横断する選択プロファイルの潜在的な地域差を探りました。本州地域全体で類似の選択プロファイルが観察されましたが、アルコール代謝に関係するADHクラスターシグナルは沖縄では比較的弱く、ALDH2遺伝子領域への自然選択は本土集団でのみ検出されました。これらの差異は、沖縄のサンプルサイズが限られているため、または選択圧が異なる可能性があるため、さらなる研究が必要です。さらに、iHS法で観察されたシグナルを検証する補完的なアプローチとしてFastSMC法を使用しました(図4B)。この方法は、過去50世代で選択の対象となった可能性がある四つの候補領域を特定しました。これには、iHS法で有意であった三つの領域(ADH、ALDH2、MHC)と、候補領域2p25.3が含まれます。これら三つの領域(ADH、ALDH2、MHC)は、以前の研究でも検出されており、日本人口における自己免疫系とアルコール代謝経路への強い選択圧の存在をさらに裏付けるものです。

A:iHS法では、自然選択された可能性のある三つのゲノム領域が特定された。
B:FastSMC法では、自然選択された可能性のある四つのゲノム領域が特定された。
グラフの横軸は染色体上の位置、縦軸は自然選択の痕跡の強さを表す。プロットは各領域の解析結果を示し、縦軸の値が大きいほど強く自然選択の影響を受けたことを意味する。
今後の期待
この研究は、日本人の起源について重要な洞察を提供しています。今まで「二重構造」モデル[16]、つまり縄文時代の狩猟採集民と大陸からの弥生時代の稲作移民の混血により現代の日本人が形成されたという説は広く受け入れられてきました。
最近日本列島の遺跡から出土した人骨のゲノムの研究による「三重構造」モデル[17]、すなわち、縄文人の祖先集団、北東アジアに起源を持ち弥生時代に日本に渡ってきた集団、そして東アジアに起源を持ち古墳時代に日本に渡ってきた集団の三集団の混血により日本人が形成されたという説が提唱されました。
しかし、先行研究で用いられた古人骨全ゲノムのサンプル数は制限されており、より多くの解析が必要と考えられていました。
本研究は、大規模な現代日本人ゲノム情報に基づいて、この三重構造モデルの裏付けになり、日本の人口構造をより適切に説明する可能性があると考えられます。さらに、本研究では初めて日本人の遺伝的構造に対する東北地方人の祖先の影響の重要性が強調されました。東北地方は歴史的に蝦夷(エミシ)が居住していた地域であり、彼らの起源を調べる必要があります。
遺伝的分析により、現生人類に非常に近い、絶滅したネアンデルタール人やデニソワ人から引き継がれたDNAが現代日本人の遺伝的多様性にどのように寄与しているか、またその病気感受性や表現型特性に与える影響はどうか、という点についての理解が深まりました。さらに、ALDH2やADHクラスターなど進化的選択を受けた特定遺伝子が日本人の免疫応答やアルコール代謝に重要な役割を果たしていることがすでに示されています。これらの遺伝特性が健康や疾患リスクにどのように影響するかについて、さらなる研究が期待されます。
この研究は、日本における遺伝学研究の新たなマイルストーンを築き、個別化医療の実現に向けた大きな一歩を踏み出すものです。まず希少遺伝変異、特定集団に特有の変異、疾患関連遺伝子の機能解析に貢献します。これらの遺伝的要素を深く理解することで、病気の原因となる遺伝要因の同定が可能になり、新たな治療法の開発ができるようになると期待されます。
さらに、日本人を含む東アジアや世界各地の人々の遺伝的起源と人類史を理解する上で重要な基礎となります。得られた知見は、日本人集団だけでなく、世界の人々に対する個別化医療の発展や新薬の発見にも役立つことが期待されます。将来の研究では、より多くのサンプルと遺伝子解析技術を活用することで、よりよい医療ソリューションの提供につながることが期待されます。









