








アビガンが今になっても承認下りない根本理由は「対ウイルス兵器製剤」だからである!!

どんな臨床試験が行われたか知っていますか
茶番劇!!
コロナなんてありません!!
国内では新型コロナウイルス感染症(COVID-19、以下、新型コロナ)の第3波流行が到来し、政府は1月7日に首都圏の1都3県に対して新型インフルエンザ等特別措置法に基づく緊急事態宣言を発令。
その後、宣言対象地域は11都府県にまで拡大し、期限を迎えた2月7日で解除されたのは栃木県のみで、残る10都府県では3月7日まで宣言が延長された。
感染者増加を阻止する手段であるワクチンは、すでに欧米で接種が始まり、日本でも今月中に医療従事者などの優先接種が開始される見込み。
しかし、すでに感染して中等症、重症となった患者での治療選択肢はまだ少ない。
現時点で使用可能な薬は、従来感染症に使われ、新型コロナの重症肺炎患者で死亡率を低下させることが報告されたステロイド薬のデキサメタゾン、アメリカでの臨床試験を基に5月に特例承認された、ウイルス増殖を抑える抗ウイルス薬のレムデシビルの2種類のみだ。
これ以外に数多くの候補薬の臨床試験が行われているが、有効性が明確に証明されているものは少ない。
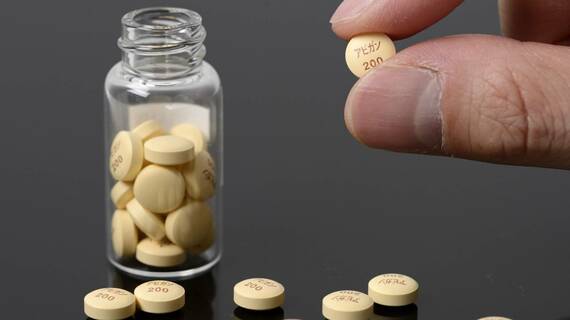
その中で一時注目を集めたのが富士フイルムの子会社・富士フイルム富山化学が開発した抗インフルエンザ薬のファビピラビル、製品名「アビガン」である。
前後編にわたって「コロナ治療薬候補」としてのアビガンについて追っていきたい。
安倍前首相が承認について強い意志を示したが…
パンデミック初期の昨年2月、中国科学院武漢ウイルス研究所はアビガンが試験管内で新型コロナウイルスに対して効果を示したと報告。
同5月には安倍晋三前首相が「今月中の承認を目指したい」とまで言及した。
当時、愛知県の藤田医科大学で医師主導の新型コロナ軽症患者に対するアビガンの臨床試験が進行中で、安倍前首相はこれで良好な結果が出れば迅速承認に結び付ける願望があったとみられる。
しかし、同7月にようやく発表された試験結果では有効性は示せなかった。
一方、富士フイルム富山化学も厚生労働省(以下、厚労省)へ新型コロナにアビガンを適応拡大するための臨床試験を実施していた。
試験開始時期が国内の第1波流行収束時に重なったことで臨床試験参加患者の確保が進まなかったが、その後の第2波流行で参加患者確保にメドがつき、最終的に良好な成績が得られたとして昨年10月に厚労省に対して製造承認を申請した。
しかし、昨年12月21日、その承認可否を審議した厚労省薬事・食品衛生審議会(薬食審)医薬品第二部会は「現時点で得られたデータから有効性を明確に判断することは困難」として、この時点での承認を見送り、継続審議とした。
承認しないのではなく、アメリカやクウェートで行われている臨床試験の結果を待って判断するとの方針を示したのだ。
国産の新型コロナ治療薬への期待が高かったためなのか、この承認見送りにはネット上などで批判が相次いでいる。
中には「厚生労働省からの天下りを受け入れない富士フイルム富山化学に対する嫌がらせ」などという陰謀論めいた言説も飛び交っている。
しかし、厚労省はその理由について、薬食審では提出されたアビガンの臨床試験が「単盲検試験」だった影響を慎重に見極めようとの意見が根強く、そのことが継続審議の大きな要因だったと説明しており、報道でも触れられている。
ただ、一般人の多くは「単盲検試験」という言葉自体初耳だろうし、一般紙の説明もややわかりにくいものが多い。
そこで「単盲検試験」とはどんなもので、今回のアビガンの承認審査でどのような影響を及ぼしたかを詳述したい。
新薬候補の臨床試験はどのように行われる?
この単盲検試験とは何かを説明するためには、新薬候補となる成分の臨床試験がどのように行われているかを知らなければならない。
まず、ある病気に対する新薬候補の製造承認を申請する際には、あらかじめヒトでの臨床試験で一定の有効性や安全性を示す必要がある。
臨床試験は第Ⅰ相試験、第Ⅱ相試験、第Ⅲ相試験と3段階に分けられているが、もっとも重要なのが最終段階の第Ⅲ相試験である。
第Ⅲ相試験では、その病気に対してこれまで最も一般的に使われていた薬(標準治療薬)あるいはプラセボ(偽薬)と新薬候補の効果や安全性を比較する必要がある。
プラセボは一般にヒトの体内に入ってもほぼ薬効も害もないもの、具体的には経口薬の場合はデンプンなどを錠剤のように成形したもの、注射薬の場合は生理食塩水が使われる。
ただ、かなり進行したがんなど、何らかの治療を行わなければ死に至る可能性が高いケースでは、無治療となるプラセボ使用は倫理上問題があるため、標準治療薬が用いられる。
そのうえで臨床試験に参加した患者を2グループに分けて、一方のグループには新薬候補、他方のグループには標準治療薬かプラセボをそれぞれ投与して、新薬候補を服用した患者で標準治療薬やプラセボを上回る効果などがあれば、そのデータを提出して審査を受けることで規制当局から製造承認を取得できる可能性が出てくることになる。
そしてこの比較のための臨床試験の実施方法には
▽二重盲検試験
▽単盲検試験
▽非盲検試験
の3種類がある。
二重盲検試験は誰に新薬候補が投与され、誰にプラセボあるいは標準治療薬が投与されているか、投与する医師、投与される患者ともに知らされていない、単盲検試験は医師だけがどの患者に何が投与されているか知っているが患者は何も知らない、非盲検試験は医師が誰に何が投与されたかを知っていて、かつ患者も自分に何が投与されたか知っている、という試験法である。
さらにこの3種類の試験方法では、それそれで2つのグループに分ける際に乱数のように無作為に分ける無作為化試験、無作為化を行っていない非無作為化試験の2種類のいずれかの方法を採用する。
つまり都合6種類の試験方法があることになるのだが、前述の3種類の試験方法については二重盲検試験、単盲検試験、非盲検試験の順、無作為化と非無作為化の間では無作為化のほうが科学的信頼度は高いと考えられている。
つまり科学的信頼度として最上位は無作為化した二重盲検試験であり、やむをえない事情がない限りは新薬の製造承認申請の臨床試験はこの方式で行われる。
厳格な科学評価を下すために
では、なぜ患者はおろか医師までも誰に何が投与されているかがわからない方法で臨床試験を行うのか?
理由は単純でバイアスを排してより厳格な科学的評価を下すためだ。
そもそも製薬企業がある新薬候補の臨床試験を行うのは、当然一定の有効性があり、将来的に市販できれば、売り上げを確保できると見込んでいるからだ。
そうである以上、臨床試験に参加する医師も患者も「今までの薬よりも有効なのではないか」という先入観を持ちがちだ。
その状態で医師があらかじめ新薬候補とプラセボがどの患者に投与されているかがわかっていれば、評価の際にバイアスが入りやすくなる。
たとえば新薬候補が投与された患者で偶然起きたにすぎない好ましい現象を「新薬候補が投与されているからだろう」と思い込んでしまう危険性がある。
患者自身が新薬候補を投与されているとわかっていると、そこから得られる安心感も影響して、医師から「具合はどうですか?」と尋ねられた際に、単なる気分で「はい、調子いいです」と答えてしまい、それが医師による薬の評価に影響を及ぼす可能性がある。
科学的な判定ではこうしたバイアスの余地をできるだけ排除しなければならない。
そのために二重盲検試験という手法が必要になる。
もっとも「医師も患者も誰に新薬が投与されているのかもわからない臨床試験なんて実施可能なのか?」と思う人もいるだろう。
結論からいえば実施可能である。
こうした臨床試験の場合、参加する医師や患者、製薬企業とは独立した「コントローラー」と呼ばれる人が存在し、このコントローラーが患者の無作為化と新薬候補やプラセボ・標準薬の割り付けを行う。
この割り付けに基づき、医師には新薬候補あるいはプラセボ・標準薬をそれとは告げられないまま渡され、そのまま患者に投与される。
そして臨床試験ではあらかじめ決められた評価項目があり、患者に投与後にこれら評価項目の変化を医師が記録して提出。
最終的に参加した患者のデータにある時点でロックがかけられて変更できない状態になる。
この後、データを集約して統計解析が行われ、それが終了して初めて医師や患者は誰に何が投与されたかが明らかにされる仕組みだ。
繰り返しになるが、今回のアビガンの臨床試験(第Ⅲ相試験)は、医師が誰に何が投与されたかを知っている単盲検試験である。
新型コロナに対するアビガンの臨床試験の概要とは?
そこで重要となるのが今回、アビガンの新型コロナに対する臨床試験の詳細である。
現在までに富士フイルム富山化学側が明らかにしている概要では、新型コロナウイルスのPCR検査で陽性となり、重篤ではないが胸部画像での肺病変(いわゆる肺炎)や37.5℃以上の発熱がある20歳から74歳までの入院患者156人が対象となっている。
これらの患者全員に一般的な肺炎治療を行いながら2つのグループに分け、1つのグループにはさらにアビガン、もう1つのグループにはプラセボをそれぞれ追加投与して効果や安全性を比較している。
ちなみに冒頭で説明したように単盲検試験であるため、医師はアビガン、プラセボがそれぞれ誰に投与されたかを知っている。
なぜベースにわざわざ標準的な肺炎治療を施し、単純にアビガンとプラセボで比較しないのかといえば、この点はまさに倫理的な問題と円滑な臨床試験の実施のためと推察される。
臨床試験対象となっているのは重い状態ではないとはいえ、すでに肺炎を起こしている新型コロナ患者である。新型コロナの場合は、時にわずか数時間で急激に重症化し、患者全体の2%、つまり50人に1人が死に至る。
このような病気でプラセボしか投与していない、すなわち実質無治療は倫理的に問題がある。
また、臨床試験実施に当たっては、参加患者から文書同意の取得が必須だが、プラセボのみ投与で無治療となる可能性があるスキームでは患者が同意をしたがらない可能性は少なくないうえ、医師側もそのような臨床試験への参加を患者に依頼しにくい。
結果として必要な参加者が集まらず、臨床試験が実施できない恐れもあるからだ。
そしてこの臨床試験では3つの評価項目を設けている。
最も優先度の高い主要評価項目は「症状(体温、酸素飽和度、胸部画像所見)の軽快かつPCR検査で陰性化するまでの期間」、これに次ぐ副次評価項目は「有害事象(副作用)」と「7段階スケールによる患者状態推移」である。
より詳しく説明すると、主要評価項目では、体温が37.4℃以下、酸素飽和度が96%以上、胸部画像所見が最悪の状態から改善の3つを患者が満たした段階にまずPCR検査を行い、この1回目のPCR検査から48時間後に2回目のPCR検査を実施する。
このPCR検査が2回とも陰性だった患者だけを選び出し、アビガンあるいはプラセボの投与開始から1回目のPCR検査陰性までの期間を比較している。
また、副次評価項目の「7段階スケールによる患者状態推移」というのは、患者の状態を退院から死亡まで7段階の状態に分類し、通常は2段階以上改善した場合を薬の効果があったと判定する手法である。
今回のアビガンの臨床試験の結果については今のところ医学論文などとして公開されていない。
ただ、富士フイルムが結果の一部を記載したプレスリリースを公表しており、主要評価項目の「症状(体温、酸素飽和度、胸部画像所見)の軽快かつPCR検査で陰性化するまでの期間」はアビガンを投与したグループが11.9日、プラセボを投与したグループが14.7日となり、アビガンを投与したグループでPCR検査による陰性化確認までの期間が約3日短くなっている。
ちなみに薬の有効性を厳格に判定する際は、統計学の計算手法で2つのグループで発生した差が偶然起きたものではない、つまりアビガンの投与かプラセボの投与かの違いにより発生したかどうか吟味する。
この手法で偶然ではない差と認められた場合は、専門用語で「統計学的に有意差が認められた」と表現する。
今回のアビガンの臨床試験では、前述の日数の差はまさに統計学的な有意差が認められている。
つまりアビガンの投与があったからこそ陰性化までの期間が約3日間短縮されたことになる。
この約3日の短縮という結果についての解釈は人によって分かれるかもしれない。
「たったの3日?」と言う人もいるだろう。
一方で患者にとっては苦痛な症状の期間が3日短くなるのは喜ばしいはずだし、現在のような感染拡大による病床逼迫を考えた場合、患者の入院期間が約3日間短縮されるならば「医療崩壊」を防ぐ可能性もある。
実はバイアスが入りやすい試験方法だった?
ところが統計学的な手法を使って厳格な科学的評価をしたうえで一定の有効性が認められたはずなのに、なぜ薬食審医薬品第二部会は、「データから有効性を明確に判断することは困難」との評価を下したのか?
ここがまさに単盲検試験であるがゆえなのだ。
くどいようだが主要評価項目を改めてすべて箇条書きにする。
(2)酸素飽和度の改善(96%以上)
(3)胸部画像所見が最悪の状態から改善
(4)PCR検査2回の陰性
(1)~(3)が満たされたうえで、(4)が満たされることが条件である。ここで医師が誰にアビガンが投与され、誰にプラセボが投与されたかを知っていることでバイアスが入る余地があるのか?
結論をいえば、(3)の胸部画像所見の判定はその可能性があるのだ。新型コロナによる肺炎の状態確認では、一般人が「レントゲン写真」と呼ぶ単純X線画像よりもCT(コンピューター断層撮影装置)画像が使われることが多い。
ただ、あくまで医師が肉眼で「ああ、肺炎の起きている面積が小さくなったな」など判定する。現在のCTでは肺炎部分の面積変化を計測して数字で表すこともできなくはないが、通常の診療ではそこまでのことは行わない。つまり厳格な科学的評価という関連からすれば、この手法はバイアスが入る余地があるということだ。
また、副次評価項目の7段階スケールの改善というのもバイアスが入りやすい。例えば、明確な死亡という状態は医師によって判定が異なるものではなく、極端な話を言えば医学的に死亡と認定された時点から一定時間が経過した状態ならば一般人でもわかる。
「退院」には医師のバイアスが入り込むことも
ところが「退院」となると、医学的に厳格な退院基準が決まっている場合ばかりでもなく、またそういう基準があったとしても、患者本人の事情などを考慮して「まだ軽度の症状はあるが、本人の強い希望もあるし、まあ退院しても問題ないだろう」という形などで医師の判断にバイアスが入り込むこともある。
つまり今回のアビガンの臨床試験は有効性に関する2つの評価項目はいずれも医師のバイアスが入る可能性が否定できないものなのである。バイアスを極力排除できたときに短縮効果が3日より短くなるのか、それとも逆に長くなるのかは現時点ではわからない。ただ、もし厳格な評価で短縮効果が3日より短くなれば、前述の「統計学的な有意差」が認められない、つまり科学的にはアビガン投与とプラセボ投与による実質無治療とに差がないという最悪の結論になる恐れもあるのだ。
ただ、原因はほかにも考えられる。後編ではアビガンには付き物の「催奇形性」の問題や審査のプロセス、海外での臨床結果などについて詳しく解説したい。

新型コロナウイルス感染症(COVID-19)の治療薬候補として一時注目を集めたのが富士フイルムの子会社・富士フイルム富山化学が開発した抗インフルエンザ薬のファビピラビル、製品名「アビガン」。
2月10日公開の「アビガンが今になっても承認下りない根本理由」に続く後編として、期待されながらもいまだ承認には至っていないアビガンの真実に迫る。
アビガンには付き物の「催奇形性」という問題
有効性評価に医師によるバイアスが入る可能性があることに加えて、今回専門家がこのデータのみで承認に同意しなかったと思われる原因はほかにも考えられる。
薬の服用は得られると思われる利益と想定されるリスクとのバランスが重要となる。やや極端な例を挙げれば、治療をしなければ死に至る可能性が高い病気に対して、服用すれば確実に1年以上は生存できるものの、服用したほとんどの人がつねに微熱や下痢に悩まされるという薬であれば、おそらく多くの患者は微熱をこらえても薬を服用することを選択するし、医師もそのほうが妥当だと判断する。
逆に命を落とすことはほとんどない病気で、苦痛な症状は時々出る程度。これに対して服用すれば症状が和らぐことになるものの、服用した患者の10人に1人に後遺症が残る副作用が出る薬があるとしたら、多くの患者は服用したがらないのではないだろうか?
実は今回のアビガンの新型コロナへの適応拡大を承認するか否かの議論には、この薬が持つ潜在的なリスクが相当程度影響していると推察される。そのリスクとは抗インフルエンザ薬としての承認時に提出された動物実験データで明らかになった「催奇形性」。端的に言えば、生殖活動期の男女や妊婦が服用した際などに生まれてくる子どもに奇形が生じる危険性である。
アビガンの動物実験ではサル、マウス、ラット、ウサギの4種類の動物で催奇形性が認められており、ラットでは初期の受精卵(初期胚)が死滅したことも報告されている。もちろん動物実験の結果と同様のことがそのままヒトで起こるとは限らない。しかし、ヒトと同じ霊長類のサルも含む4種類の動物すべてで催奇形性が確認される以上、ヒトでも十分起こりうると考えるのが妥当である。
実際、アビガンの抗インフルエンザ薬としての承認時はこの点が最大の論点となった。企業側は季節性インフルエンザの治療薬として承認申請を求めていたが、シーズンごとに確定患者だけで数十万人規模となる季節性インフルエンザに催奇形性があるアビガンを承認することに厚労省側が難色を示したのだ。
ただ、新型インフルエンザのパンデミック後のことで、その当時まで承認されていた抗インフルエンザ薬とは異なる新しい作用の仕方を示すということもあり、苦肉の策として「新型または再興型インフルエンザウイルス感染症(ただし、ほかの抗インフルエンザウイルス薬が無効または効果不十分なものに限る)」と極めて限定された適応で承認された。
しかも、新型インフルエンザのパンデミック発生時に国が出荷の可否を決めるという、これまた前代未聞の条件が付き、今回の新型コロナパンデミック以前は新型インフルエンザ対策用の国の備蓄のため以外では製造されておらず、一般の医療機関には在庫すらなかったほどだ。
さらに新型インフルエンザでの流通であっても厳格管理が前提となっており、妊婦や妊娠の可能性のある女性への投与の回避のほか、妊娠するあるいはさせる可能性がある男女では投与中および投与後7日間の避妊の徹底が求められている。
つまるところ新型コロナの症状改善までの期間は約3日短縮できるかもしれないが、そこにはバイアスが含まれている可能性があり、一方で副作用により次世代にまでわたって害を及ぼす可能性があるというのが今のアビガンの置かれた現実である。そうである以上、審査側として慎重になるのは当然と言えるだろう。
「審査機関に提示して合意を得たもの」?
今回の継続審議が明らかになった時点で、単盲検試験という手法について富士フイルム富山化学が「審査機関に提示して合意を得たもの」と、やや恨みがましいとも解釈できるコメントをしていることが一部で報じられている。
審査機関とは、製薬企業が新薬の製造承認を申請した際に提出したデータの1次的審査をする厚労省所管の独立行政法人「医薬品医療機器総合機構(PMDA)」のことである。ここでの審査が審査報告書としてまとめられて外部の専門家で構成される厚労省薬食審に提出され、最終的な承認可否が判断される。今回のアビガンの場合、この最終段階の薬食審で物言いがつき、承認されないままになっている。
PMDAは新薬候補の1次的な審査にとどまらず、製薬企業が新薬候補の動物実験やヒトでの臨床試験を行う際の法令への適合や科学的妥当性などを助言する事前相談業務も行っている。今回のような緊急性の高い医薬品の開発に際して製薬企業はほぼ確実にPMDAに事前相談するはずであり、富士フイルム富山化学のコメントにあるPMDAがアビガンの臨床試験を単盲検試験で行うことに同意していたのは事前相談でのことだろう。
ネット上では「事前に臨床試験のやり方に同意していながら、お役所は富士フイルムに嫌がらせをしている」などの陰謀論めいた言説を目にすることもあるが、PMDAが単盲検試験の実施に同意していたのは承認審査を遅らせるためではなく、逆にアビガンの新薬承認をスピーディーに行う意図があったと考えるほうが妥当だ。そう考えられるのは主に3つの理由からだ。
1つ目には感染症の場合、ほかの病気と違って新薬候補の臨床試験実施に独特の難しさがあるからだ。例えば糖尿病や高血圧のような病気で新薬候補の臨床試験を行う場合、参加患者の確保は比較的容易である。これらの病気ではつねに一定数の患者が医療機関を継続的に受診しているため、参加打診をする患者を見つけやすい。
ところが感染症はいつどこで患者が発生するかはまったく不明だ。しかも、感染症の蔓延は今回のように社会全体を混乱させるので、感染者の発生を防止しようとするさまざまな対策が動いている。この結果、構造的に臨床試験実施の前提になる参加患者の確保が容易ではない。
モグラたたきに例えると、糖尿病や高血圧の新薬候補の臨床試験参加者確保は、つねにどの穴からどのタイミングでモグラが出てくるかがわかっているモグラたたき、感染症でのそれはどの穴からどのタイミングでモグラが出てくるかはわからないモグラたたきである。
命を落とす可能性があるのに参加できるか
2つ目にはまさに命を落とす可能性がある感染症にかかっている患者が、自分にどんな治療を行われているかがわからない二重盲検試験への参加を求められれば参加を躊躇する可能性がある。それでは臨床試験そのものが成り立たない。
3つ目には、中等症から数時間で一気に重症化することがある新型コロナの特性を考えると、臨床試験に参加する医師側も単盲検試験のほうが好都合という可能性もある。というのも、臨床試験に参加中の患者で急速な重症化が起きた場合、医師は最短で最適な対処法を模索するため、患者にアビガンとプラセボのどちらが投与されていたかを知る必要がある。
この際に二重盲検試験ならば、医師は容体が急変している患者を前に前述のコントローラーに何とかアクセスして確認するというまどろっこしいプロセスが必要になる。これに対し単盲検試験ならば、その場で迅速に対処方針を決定できる。
このような3つのハードルを考え、臨床試験の早期・円滑な実施を念頭にPMDAが富士フイルム富山化学側の提示した単盲検試験に同意したこと自体、何ら不思議はない。また、PMDAに対する事前相談の段階で今回最終判断のために待っているアメリカとクウェートでの二重盲検試験の計画は富士フイルム富山化学から通知されていたはずである(していないということはまずありえない)。
このためPMDA側も単盲検試験という、とりあえず走り出しやすいスキームで先行し、そこで極めて良好な結果が得られた場合はその段階で、そうでなかった場合は二重盲検試験の結果を待って薬食審が適切に判断してくれるだろうという腹積もりがあったと思われる。
そして最終的に確定した単盲検試験結果は約3日短縮という微妙な差だったため、バイアス混入の是非が真剣味を帯びてきたと考えられる。その意味ではPMDAが単盲検試験という手法に事前に同意したことと、今回の薬食審での承認先送りはまったく矛盾しない。
すでに一部が明らかになった海外での臨床試験結果
いずれにせよアメリカ、クウェートでの臨床試験の結果次第なわけだが、実はクウェートでの二重盲検試験は終了し、1月27日に結果の一部が明らかになった。試験は富士フイルム側が中国、ロシアを除くアビガンの製造販売ライセンスを供与しているインドの製薬大手ドクター・レディーズ・ラボラトリーズが実施したものだ。
試験では中等症から重症で入院中の新型コロナ患者353人を対象として症状改善までの時間を評価したが、全体では症状改善までの期間が、アビガンを投与されたグループとプラセボを投与されたグループでは統計学的有意差が認められなかった、ありていに言えばアビガンの有効性は証明できなかった。
ただ、参加患者をよりリスクの低い患者181人のみに絞って解析すると、アビガンを投与されたグループではプラセボを投与されたグループに比べて退院までの期間が3日短縮し、この差では統計学的に有意な差が認められたという。
やや突っ込んで解釈を示すならば、クウェートでの二重盲検試験では、よりリスクの低い患者、おそらく中等症の患者で、医師のバイアスが入りやすい「退院」という基準で3日間短縮効果が認められたということ。率直に言ってかなり微妙な結果と言える。この点を併せて見ても、現時点で厚労省の薬食審が承認を先送りしていることはとくに不自然なことではなく、この薬の偽らざる実力の結果である。
残るはやはりドクター・レディーズ・ラボラトリーズが、アメリカで軽症から中等症者を対象に症状改善や入院・ICU入りリスクの軽減を評価する目的で行っている二重盲検試験の結果がどうなるかだ。もっとも、新型コロナの軽症者はほぼ無治療でも回復するのが常識で、この試験で劇的な効果は示せないのではないかとの見方もある。
ここまで
奴らは最早なりふり構わず「何でもあり!!」と、昆虫食、そして猛毒蚊に人々を襲わせる準備をしています!!
腑抜の人々は、奴らの言いなりです!!
自分どころか家族さえも、守りません!!
-
新型コロナを証明した論文はありません!!
公的機関のコロナ情報はすべて嘘です!!
新型コロナは存在しません!!
ワクチンには予防効果はありません
発症効果を防ぐ効果もありません!!
重症化を防ぐ効果もありません!!
ワクチンの中身の正体は
酸化グラフェンによる血栓と、M-RNA修飾ウリジンを使った遺伝子組み換えで免疫破壊
この二つを主体に貴方の身体を徹底的に機械する遺伝子兵器です。
-
ワクチンや食品に含まれる社会毒を無毒化する
ビタミンB2・ビタミンC・ビタミンE・コエンザイムQ10
納豆・ニンニク・ゆで卵・ゴーヤ・トマト・ブロッコリー
きのこ・梅干し・胡麻ナッッ・オリーブオイル・寒天
-
非加熱の塩
nRNAワクチン汚染は、想像以上に深刻
新型コロナウイルスが存在することを証明した人は世界に誰一人としていません!!
それを政府も厚生労働省も「新型コロナウイルス」と呼びます!
病原体を説明することが出来ないものに対してワクチンを作れません!!
遺伝子の確認も病原体を証明しないとPCR検査も作れない!!








