がんの予防や治療における漢方治療の存在意義を考察しています。がん治療に役立つ情報も紹介しています。
「漢方がん治療」を考える
カレンダー
| 2024年12月 | ||||||||
| 日 | 月 | 火 | 水 | 木 | 金 | 土 | ||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | ||
| 8 | 9 | 10 | 11 | 12 | 13 | 14 | ||
| 15 | 16 | 17 | 18 | 19 | 20 | 21 | ||
| 22 | 23 | 24 | 25 | 26 | 27 | 28 | ||
| 29 | 30 | 31 | ||||||
|
||||||||
goo ブログ
過去の記事
カテゴリ
最新の投稿
最新のコメント
最新のトラックバック
ブックマーク
プロフィール
検索
gooおすすめリンク



| URLをメールで送信する | |
| (for PC & MOBILE) | |
693)免疫原性細胞死の誘導(その1): 2-デオキシ-D-グルコース

図:抗がん剤や放射線治療などによってがん細胞が死滅すると(①)、死滅したがん細胞からがん抗原が放出される(②)。がん細胞から放出されたがん抗原は未熟樹状細胞に取り込まれ、未成熟樹状細胞は活性化されて成熟樹状細胞に分化誘導される(③)。成熟樹状細胞は最寄りのリンパ節に移動し、MHC(Major Histocompatibility Complex)のクラスI及びクラスIIに結合したがん抗原をTCR(T細胞受容体)を介して、CD4+T細胞(ヘルパーT細胞)とCD8+T細胞(キラーT細胞)に提示する(④)。がん抗原に反応するキラーT細胞(細胞傷害性T細胞)は抗原提示によって活性化されてクローナルに増殖し(⑤)、がん細胞をがん抗原特異的に攻撃する(⑥)。抗がん剤や放射線治療に2-デオキシ-D-グルコースを併用すると、死滅したがん細胞の免疫原性を高めた免疫原性細胞死を誘導できる(⑦)。
693)免疫原性細胞死の誘導(その1): 2-デオキシ-D-グルコース
【体にはがん細胞を排除する免疫力が備わっている】
「免疫」というのは「疫病(伝染病・感染症)」から「免れる」というのが原義です。
免疫は「二度なし現象」ともいわれます。これは一度感染した病原体には二度と感染しない、もしくは感染しても軽症ですむという意味です。
ヒトに対する病原性が弱い牛の天然痘(牛痘)を接種して、人間の天然痘の感染を予防するワクチン療法を確立したエドワード・ジェンナー(Edward Jenner)は、「免疫学の父」と言われています。
ジェンナーが初めて種痘(牛痘)を接種したのは1796年ですが、人間の天然痘(人痘)の膿を乾燥させてウイルスを不活性化して人間に接種する方法は、インドでは紀元前から行われていたようです。乾燥してウイルスを不活性化した人痘を使った天然痘の予防法はインドから中国に伝わり広く行われました。日本でもジェンナーの種痘接種より早く、現在の福岡県にあった秋月藩の藩医の緒方春朔が、1792年人痘種痘法を実施し成功させています。
ジェンナーの牛痘を使った種痘は生ワクチンで、インドの人痘を使った方法は死活化ワクチンに相当します。
このように、人類はかなり昔(少なくとも2000年以上前)から感染症における「二度なし現象」に気づいており、感染症の予防に利用してきました。
免疫系は自己と非自己を識別し、非自己を排除する生体防御システムです。
異物(非自己)を排除する免疫系は、自己の変異細胞であるがん細胞も排除して生体を防御するという「がん免疫監視説(cancer immunosurveillance)」の概念をフランク・マクファーレン・バーネット(Frank Macfarlane Burnet)が1950年代から提唱し、1960年代には広く認められるようになりました。
バーネットは、免疫寛容とクローン選択の概念を唱え、1960年にノーベル医学生理学賞を受賞したオーストラリアの免疫学者です。
免疫力の低下ががんの発生や進行を促進することは多くの証拠があります。
免疫抑制剤を使用している臓器移植患者や、HIV感染(エイズ)などによる免疫不全状態の患者はがんの発生率が高いことが知られています。
免疫機能の低下の原因として最も重要なのは老化によるものであり、そのほか精神的・肉体的なストレスや栄養障害なども重要です。老化とともにがんの発生が増えることや、ストレスががんの発生や進行を促進することも、その原因は免疫力が低下するからです。
人間の免疫力は18~22才くらいをピークにして年令とともに衰え、がん年令の始まりといわれる40才台の免疫力はピーク時の半分まで下がり、その後も加齢とともに下降するといわれています。
免疫力を高めてがんの消滅を目的とする「免疫療法」は、手術、化学療法、放射線療法に次ぐ第4のがん治療法として重要視されています。
免疫療法は、19世紀末に外科医であるW.B.コーリーが細菌由来毒素であるコーリートキシン(Coley Toxin)をがん患者に投与して、免疫を賦活させることによりがんを治癒させたことに端を発します。
溶連菌製剤のピシバニールや、カワラタケの菌糸体から見つかった蛋白結合多糖(ベータグルカンに蛋白が結合)のクレスチンなど、免疫系を活性化する目的のがん治療薬もあります。
最近では、オプジーボなどの免疫チェックポイント阻害剤で、一部のがん患者において、がんが縮小したり消滅することが明らかになっています。
つまり、体に備わった免疫監視機構を十分に高めることができれば、がん細胞を死滅させ、がんを縮小したり、消滅させることもできるのです。
しかし現実的には、免疫療法の効果はまだ弱いと言わざるをえません。
その理由の一つとして、がん組織にはがん細胞を攻撃するエフェクター細胞(キラーT細胞やナチュラルキラー細胞など)の働きを阻害する細胞(骨髄由来抑制細胞、制御性T細胞、M2型腫瘍関連マクロファージなど)や因子(プロスタグランジンE2やキヌレニンや乳酸など)の存在があります。
これに対する代替療法的な対処法に関しては前回(692話)紹介しています。
「抗腫瘍免疫を抑制している要因の排除」に加えて、「エフェクター細胞の活性を高める方法」、「がん抗原の発現や認識を高める方法」などを組み合わせると、さらに抗腫瘍免疫を高めることができます。

図:がん細胞から放出されるがん抗原を樹状細胞が取り込むと(①)、活性化された樹状細胞は1型ヘルパーT細胞(Th1細胞)を活性化する(②)。Th1細胞はIL-2やIFN-γの産生を亢進し、キラーT細胞の活性を高め(③)、がん細胞に対する細胞性免疫を増強する(④)。しかし、がん組織内には骨髄由来抑制細胞(MDSC:Myeloid-derived suppressor cell)、制御性T細胞(Treg:Regulatory T cell)、腫瘍関連マクロファージ(TAM:Tumor-associated macrophage)などの細胞が集積し、これらの細胞はキラーT細胞の働きを抑制している(⑤)。がん組織から産生されるIL-10やVEGFやTGF-βやプロスタグランジンE2(PGE2)などはキラー細胞の活性を抑制し、骨髄由来抑制細胞を動員するなど、抗腫瘍免疫の抑制に関与する(⑥)。
【がん細胞は免疫原性を弱めて免疫監視機構から逃れている】
免疫細胞が体内を監視していて、異常を起こした細胞を見つけて排除しているという「がん免疫監視説(cancer immunosurveillance)」を1950年代にバーネットが提唱しました。この仮説が受け入れられなかった時期もありますが、現在では「がん免疫監視説」は仮説ではなく定説と考えられています。
がん免疫の存在を実証した実験としてマウスの線維肉腫発がん実験があります。発がん剤により誘導した線維肉腫を切除した後に、切除した腫瘍をその元のマウスに移植すると拒絶されますが、正常のマウス(ナイーブマウス)では線維肉腫細胞は生着し、増殖します。
しかし拒絶したマウスの脾臓から取り出したCD8+ T細胞(キラーT細胞)を移入したナイーブマウスでは、移植した線維肉腫細胞を拒絶します(肉腫が増殖しない)。これはCD8+ T細胞による腫瘍拒絶(がん免疫)の存在を示しています。
つまり、CD8+ T細胞が認識するがん抗原の同定とがんワクチンの開発の現実性を示唆しています。
私たちの体内では、毎日多数のがん細胞ができていますが、免疫監視の機能が正常であれば、通常はがん細胞が増殖して成長することは無いのです。
しかし現実は、発生したがん細胞は増殖し、転移しています。それには大きく分けて2つの理由があります。
一つは「免疫監視機構」の機能が低下して、がん細胞を排除できないという理由です。これにはエフェクター細胞(キラーT細胞やNK細胞など)の機能低下や、免疫抑制性の要因(制御性T細胞や骨髄由来抑制細胞など)の存在などが含まれます。
もう一つは、がん細胞が抗原性を低下させているという理由です。がん細胞は不均一な集団で、がん抗原の発現の低いがん細胞が免疫監視機構による排除を回避して選択的に生き残る可能性があります。がん抗原の発現が低下すれば、免疫細胞は正常細胞と区別できないので、そのがん細胞を排除できません。
がん免疫監視機構が存在するにもかかわらず、がん細胞が発生し進行する理由を米国ワシントン大学医学部のR.D. Schreiberらは「がん免疫編集説(cancer immunoediting)」で説明しています。
この「がん免疫編集説」では、発がんにおける免疫系とがんの関わりは「排除相(Elimination)」「平衡相(equilibrium)」「逃避相(escape)」とよばれる3 相に分けられます。
最初に出現した変異細胞(がん細胞)は免疫原性が高いため、免疫監視機構によって異物と判断され、免疫担当細胞が攻撃することによって排除されます(排除相)。
しかし免疫原性の低い(免疫応答が容易に誘導されるがん抗原を有しない)がん細胞は免疫担当細胞からの攻撃にさらされないため、排除されることなく長期にわたって選択的に生存します(平衡相)。
さらに、がん組織内に免疫抑制性の細胞(骨髄由来抑制細胞や制御性T細胞など)や因子(プロスタグラジンE2や組織の酸性化など)が増え、積極的に抗腫瘍免疫応答を抑制する環境を作り上げ、免疫系からの攻撃を逃避することで無限に増殖し臨床的がんになります(逃避相)。
つまり、がん免疫編集説(Cancer Immunoediting)に従えば、がん細胞は免疫系からの攻撃を受けにくい免疫原性の低いがん細胞を選択する(免疫選択)とともに、生体に備わっている様々な免疫抑制機構を用いて免疫系から逃避(免疫逃避)することで、 生体内で増殖し臨床的な「がん」となるということです。
したがって、がん免疫療法を成功させるには,がん細胞によって構築された免疫抑制ネットワークを解除すると共に,免疫原性の低いがん細胞に対して強力な免疫応答を誘導する必要があります。
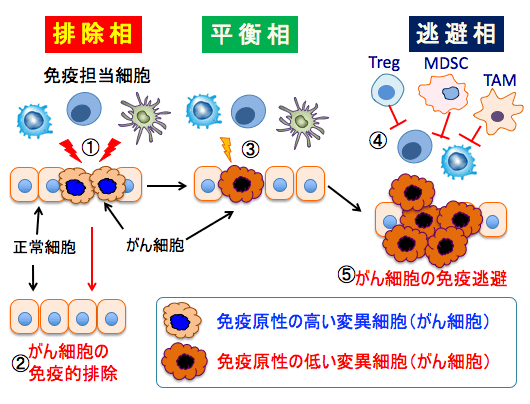
図:がん細胞が発生すると、がん細胞に存在するがん抗原をターゲットにして樹状細胞やマクロファージやキラーT細胞などの免疫担当細胞(エフェキター細胞)ががん細胞を攻撃し(①)。がん細胞は免疫的に排除される(②)。がん抗原を多く発現している「免疫原性の高い変異細胞(がん細胞)」はこの排除相で排除されるが、がん抗原の発現の弱い「免疫原性の低い変異細胞(がん細胞)」は免疫担当細胞からの攻撃が弱いので、排除されることなく長期にわたって選択的に生存する(③)。この状態を平衡相と言う。がん細胞を攻撃するエフェクター細胞の働きを阻害する骨髄由来抑制細胞(MDSC)や制御性T細胞(Treg)やM2型腫瘍関連マクロファージ(TAM)や免疫阻害因子(プロスタグランジンE2やキヌレニンや乳酸など)によってエフェクター細胞の機能が低下すると、がん細胞は免疫監視から逃避して増殖し、臨床的がんになる(⑤)。これを逃避相と言う。がん細胞は抗腫瘍免疫応答を抑制する環境を積極的に構築している。
【がん細胞は多数のがん抗原を持っている】
がん細胞は、遺伝子の突然変異によって正常な増殖制御を失うことで発生します。さらに、がんが進行する過程で、ゲノムの不安定性に基づく遺伝子変異を蓄積します。
これらの遺伝子変異は正常とは異なる変異タンパク質を作ります。この変異タンパク質は免疫系に「非自己」として認識され、免疫応答の標的として免疫反応を強く誘導する抗原となります。このような抗原をネオアンチゲン(neoantigen)と言います。
ネオアンチゲンはがん細胞の遺伝子変異の結果,アミノ酸が置き換わって新規に生じた抗原で、もともとの宿主体内には存在しなかった抗原であるため、がん細胞を排除するキラーT細胞のターゲットになります。つまり、ネオアンチゲンはがんワクチンの候補となります。
免疫系は正常な「自己」の抗原には反応しませんが、ネオアンチゲンは正常な細胞には存在しないため「非自己」として認識されて強い免疫反応の標的になるのです。
一方で、遺伝子変異は個々の患者で異なり共通するものはごくわずかであるために、患者ごとの個別対応が必要なことも分かってきました。
最近は、遺伝子の塩基配列を高速に読み出せる次世代シーケンサー(DNA解析装置)の登場により、個々の患者のがん細胞で生じている遺伝子変異を迅速かつ正確に解析できるようになりました。
つまり、患者のがん細胞に生じた遺伝子変異の中から、その患者の免疫反応を強く誘導するネオアンチゲンを比較的簡単に見出すことが可能になりました。
このような個別のネオアンチゲンを標的としたがんワクチン療法も可能になっています。
しかし、個別のネオアンチゲンを見つけてがんワクチンを作るという治療には限界もあると思います。
先ほどの「がん免疫編集説」では、排除相(elimination)では生体内に生じた変異細胞(がん細胞)が種々の免疫細胞により排除されますが、排除相で排除しきれなかった免疫原性の低い(免疫応答が起こりにくい)変異細胞が生き残ります。つまり、免疫応答が起こりにくいがん細胞が生き残るように免疫系により編集(edition)を受けているのです。
これは抗がん剤治療と同じです。抗がん剤投与によってその抗がん剤に感受性のあるがん細胞は死滅しますが、その抗がん剤に耐性のがん細胞が選択的に生き残ります。
同様にネオアンチゲンを特定してがんワクチンを作っても、いずれはそのネオアンチゲンを持たないがん細胞が生き残って、免疫監視から逃避します。
風邪に対するワクチン療法が成功しないのは、風邪を引き起こすウイルスの種類が多いからです。また、インフルエンザも変異すると既存のワクチンが効かなくなります。
そこで、がん細胞に存在する(あるいは治療に伴って新たに出現する)多様ながん抗原に生体内で免疫細胞が作用する環境を整える方が治療戦略としては合理的だと思います。
つまり、「抗腫瘍免疫応答を抑制するメカニズムや微小環境の改善」、「がん細胞を攻撃するエフェクター細胞の活性化」、「がん抗原の認識(抗原性)を高める」という3つを同時に行えば、多様ながん抗原をターゲットにしたがん免疫療法が実践できます。
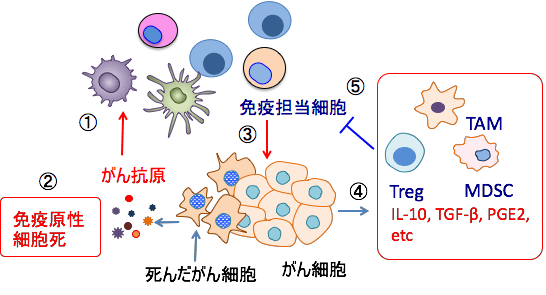
図:がん細胞が死滅するとがん抗原が放出され、樹状細胞を活性化し、さらにキラーT細胞などがん細胞を攻撃する免疫担当細胞の活性を高める(①)。このとき、免疫原性細胞死を誘導するとがん抗原の認識を高めることができる(②)。免疫担当細胞はがん細胞を死滅させ排除するように働く(③)。がん組織から炎症性サイトカインやケモカインなどが産生され(④)、制御性T細胞(Treg)や骨髄由来抑制細胞(MDSC)や腫瘍関連マクロファージ(TAM)などが動員され、免疫担当細胞の働きを抑制する(⑤)。したがって、がん免疫療法を成功させるには,免疫担当細胞の活性を高め、がん細胞によって構築された免疫抑制ネットワークを解除すると共に、免疫原性の低いがん細胞に対して強力な免疫応答を誘導する必要がある。
例えば、ビタミンやミネラルやタンパク質の欠如は免疫担当細胞の働きを低下させます。したがって、栄養状態を良くすることはがん免疫治療において重要です。
がん細胞は抗腫瘍免疫応答を抑制する微小環境を積極的に作り上げています。この免疫抑制ネットワークの解除ができなければ、がん免疫療法は成功しません。
がん細胞の解糖系を抑制し、ミトコンドリアが活性化して、がん組織の酸性化を軽減するだけで、免疫担当細胞の活性が高まります。
がん細胞のがん抗原の発現を増やすことは困難ですが、がん抗原の認識と免疫応答を増強することは「免疫原性細胞死の誘導」という方法で達成できます。
【ダメージ関連分子パターンが免疫応答を刺激する】
体を構成する正常細胞は毎日多数の細胞がアポトーシスで死滅し、組織幹細胞が細胞分裂して組織の細胞を供給しています。
このような生理的な死に対して、体がいちいち反応して炎症や免疫応答を行えば、大変なことになります。しかし、このような生理的な細胞死は、炎症や免疫応答を引き起こさない死に方をするので、問題は起こりません。
一方、何らかのダメージやストレスで細胞が傷害されたときは、それを認識して対応する必要があります。例えば、神経が熱や痛みを感じるようになっているのは、体に危害を与える傷害を認識してそれを避ける必要があるからです。
同様に、細胞がダメージを受けたとき、そのような細胞からは通常であれば細胞内に隠れている成分が放出され、炎症細胞や免疫細胞を活性化するメカニズムが存在します。
このような炎症を引き起こす細胞内にある成分をDAMPs(damage-associated molecular patterns; ダメージ関連分子パターン)と総称しています。
細胞傷害に伴って細胞から放出され、周囲の組織や細胞に危険を知らせるアラームのような役割を担う因子のことです。
DAMPsが細胞外や細胞膜上に露出するような細胞死が起こると、炎症細胞(マクロファージや好中球など)やリンパ球や線維芽細胞などが動員され、炎症反応が引き起こされ、ダメージを受けた組織の修復が起こります。
このメカニズムは自己免疫疾患などの慢性炎症性疾患の原因ともなります。
しかし、抗がんや放射線を使ったがん治療の場合は、このダメージ関連分子パターン(DAMPs)を誘導する細胞死のメカニズムを利用すると、がん特異免疫を増強できることが知られています。
DAMPsは骨髄や末梢組織から未成熟な樹状細胞をがん組織に動員し、樹状細胞は死滅したがん細胞から放出されたがん抗原によって活性化され成熟します。樹状細胞から抗原提示を受けて活性化したキラーT細胞(細胞傷害性T細胞)は増殖してがん細胞を抗原特異的に攻撃します(下図)。

図:抗がん剤(①)によってがん細胞が死滅すると、死滅したがん細胞からカルレチキュリン(CRT)やATPやHMGB1(High-mobility group box 1 protein)などのダメージ関連分子パターン(damage-associated molecular patterns ; DAMPs)が放出される(②)。DAMPsは骨髄や末梢組織から未成熟な樹状細胞をがん組織に動員する(③)。がん組織において死滅したがん細胞から放出されたがん抗原は未熟樹状細胞に取り込まれ、未成熟樹状細胞は活性化されて成熟樹状細胞に分化誘導される(④)。成熟樹状細胞は最寄りのリンパ節に移動し、MHC(Major Histocompatibility Complex)のクラスI及びクラスIIに結合したがん抗原をTCR(T細胞受容体)を介して、CD4+T細胞(ヘルパーT細胞)とCD8+T細胞(キラーT細胞)に提示する(⑤)。がん抗原に反応するキラーT細胞(細胞傷害性T細胞)は抗原提示によって活性化されてクローナルに増殖し(⑥)、がん細胞をがん抗原特異的に攻撃する(⑦)。
放射線照射や一部の抗がん剤が免疫原性の高い細胞死を誘導することが知られており、このような細胞死をもっと効率的に行う手段があれば、がん治療の効果を高めることができます。
DAMPsは、細胞質や核やミトコンドリアや小胞体などに存在する成分が放出されたもので、炎症細胞や免疫細胞を刺激します。
DAMPsとしては、ミトコンドリアのATP、 DNA、フォルミルペプチド、核のヒストンやHigh-mobility group box 1 protein(HMGB1)、High-mobility group nucleosome binding protein 1(HMGN1)、細胞質のATPやF-アクチン、小胞体のカルレチキュリン(Calreticulin)などが知られています。(下図)
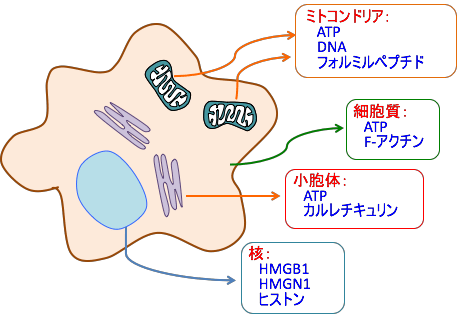
図:細胞がダメージを受けたとき、通常であれば細胞内に隠れていた成分が放出され、炎症細胞や免疫細胞を活性化するメカニズムが存在する。このような炎症を引き起こす細胞内にある成分をDAMPs(damage-associated molecular patterns; ダメージ関連分子パターン)と総称している。DAMPsは、細胞質や核やミトコンドリアや小胞体などに存在する成分が放出されたもので、炎症細胞や免疫細胞を刺激する。DAMPsとして、ミトコンドリアのATP、DNA、フォルミルペプチド、核のヒストンやHigh-mobility group box 1 protein(HMGB1)、High-mobility group nucleosome binding protein 1(HMGN1)、細胞質のATPやF-アクチン、小胞体のカルレチキュリン(Calreticulin)などが知られている。
抗がん剤治療が免疫原性細胞死を誘導することが知られています。その作用は、抗がん剤の種類や投与量によって異なるようです。
免疫原性細胞死を誘導できる抗がん剤として多くのアントラサイクリン系薬(doxorubicin, epirubicin, idarubicinなど)、mitoxantrone、 オキサリプラチン(oxaliplatin)、シクロフォスファミド(cyclophosphamide) 、bortezomibなどが報告されています。
例えば、大腸がんや乳がんでは、アントラサイクリンやオキサリプラチンががん組織内のキラーT細胞の数を増やし、免疫抑制性に作用する制御性T細胞の数を減らすことが報告されています。
従来、抗がん剤の抗腫瘍効果は、がん細胞を直接死滅させる機序によると考えられてきました。しかし、免疫応答を誘導して、免疫監視機構を活性化する機序も関与していることが指摘されるようになったのです。
したがって、抗がん剤治療と免疫療法を積極的に併用する有用性も根拠があります。
【抗がん剤はがん細胞の免疫原性細胞死を誘導する】
抗がん剤はがん細胞を死滅して、抗腫瘍免疫を誘導する作用があることが指摘されています。免疫療法を行うときに抗がん剤をうまく利用すると、抗腫瘍免疫を増強できるという考えです。 以下のような論文があります。
Immunogenic effects of chemotherapy-induced tumor cell death.(化学療法による腫瘍細胞死の免疫原性効果)Genes Dis. 2018 May 17;5(3):194-203.
【要旨】
従来の化学療法の臨床的効果が腫瘍細胞への毒性のみに起因するのではなく、免疫監視機構の活性化にも起因することが、最近の多くの動物実験や臨床研究によって示唆されている。抗がん剤による免疫監視機構の活性化というメカニズムは過去の研究においてほとんど無視されてきた。
抗腫瘍免疫応答は、免疫原性細胞死(immunogenic cell death)が引き金になって誘導される。
この免疫原性細胞死は、カルレチキュリン(calreticulin)の細胞表面移行、ATPおよびhigh mobility group box 1 (HMGB1)タンパク質の細胞外放出、タイプ1のインターフェロン応答の刺激によって特徴付けられる細胞死である。
ここでは、従来の化学療法剤が免疫原性細胞死誘導剤として作用し、免疫抑制性の微小環境内で腫瘍浸潤リンパ球の働きを調節し、抗腫瘍免疫を再活性化できることを示す最近の研究を要約する。
通常の化学療法のこのような免疫学的効果は、がん患者の予後をより良くする可能性がある。さらに、免疫原性細胞死を誘発する化学療法と免疫療法との組み合わせは、がん患者の臨床転帰を改善するための有望なアプローチである。
免疫細胞にはがん細胞を攻撃するキラーT細胞やナチュラルキラー(NK)細胞だけでなく、免疫システムを抑制する制御性T細胞、骨髄由来抑制細胞、M2型腫瘍関連マクロファージなどがあります。
免疫を抑制するメカニズムは、T細胞の暴走による正常細胞での攻撃を避けるために存在します。しかし、がん細胞はこのような免疫抑制性のメカニズムを利用して、T細胞からの攻撃を回避しています。
免疫チェックポイント阻害剤などで、がん細胞の免疫監視機構回避のメカニズムを阻止できれば、がん細胞を免疫の力で排除できます。その結果、転移がんでもがんを根治できる可能性が出てきます。
【2-デオキシ-D-グルコースは解糖を阻害する】
2-デオキシ-D-グルコース(2-Deoxy-D-Glucose:2-DG)はグルコース(ブドウ糖)の2位のOHがHに変わっているグルコース類縁物質です(下図)。

2-DGはグルコースと同じトランスポーター(輸送担体)で取り込まれるので、グルコースの取り込みが亢進しているがん細胞に多く取り込まれます。
細胞内では、ヘキソキナーゼによってリン酸化されて、2-デオキシグルコース-6リン酸(2-DG-6リン酸)に変換されますが、この2-DG-6リン酸は解糖系の先の代謝系には進めない(ヘキソキナーゼの先の解糖系酵素で代謝できない)ので、細胞内に蓄積します。
特に、がん細胞は2-DG-6リン酸を脱リン酸化して2-DGに戻すホスファターゼ活性が低下しているので、がん細胞内に2-DG-6リン酸が蓄積します。
蓄積した2-DG-6リン酸は解糖系酵素のヘキソキナーゼとホスホグルコースイソメラーゼ(グルコースリン酸イソメラーゼ)をフィードバックで阻害する作用があり、取り込まれたグルコースの解糖系での代謝を阻害し、ATP産生や物質合成を低下させます。

図:2-デオキシ-D-グルコース(2-DG)はグルコース(ブドウ糖)の2位のOHがHに変わっているグルコース類縁物質(①)で、グルコースと同様にグルコーストランスポーター(GLUT1)によって細胞内に取り込まれる(②)。細胞内のヘキソキナーゼで2-DG-6リン酸(2-DG-6-PO4)になるが、それから先の解糖系酵素では代謝できないので細胞内に蓄積する(③)。蓄積した2-DG-6リン酸はヘキソキナーゼ(HK)とホスホグルコースイソメラーゼ(PGI)をフィードバック的に阻害するので、グルコースの解糖系での代謝を阻害する(④)。
【2-DGはペントースリン酸経路を阻害する】
解糖中間体は多くの生合成系へと流れていきますが、その一つがペントースリン酸経路です。
ペントースリン酸経路とは、解糖系の中間体のグルコース6リン酸から分岐し、同じく解糖系の中間体 グリセルアルデヒド3リン酸に戻る経路(回路)です。解糖系と同様に細胞質に存在する経路で、補酵素の一つであるNADPHを産生し、核酸の原料となるリボース5リン酸などの5単糖 (ペントース) を産生します。
NADPHは還元剤です。脂肪酸やステロイドの合成、抗酸化物質のグルタチオンやチオレドキシンの還元剤として使用されます。
解糖系はATPを産生します。ペントースリン酸経路はATP産生には関与せず、核酸の原料や還元剤(NADPH)の産生を行っています。
細胞が増殖するにはエネルギー(ATP)だけでなく、核酸や脂肪酸などの物質合成や、酸化ストレスを軽減する還元剤の需要も増えます。したがって、がん細胞では、解糖系とペントースリン酸経路での代謝が亢進しています。
2-デオキシ-D-グルコースはヘキソキナーゼとホスホグルコースイソメラーゼを阻害するので、グルコースの解糖系と同時にペントースリン酸経路を阻害してNADPHと5単糖 (ペントース)の産生を阻害します(下図)。

図:解糖系は1分子のグルコースが2分子のピルビン酸に分解される過程で2分子のATPが産生される(①)。グルコース6リン酸から派生するペントースリン酸経路(②)では、還元剤のNADPHが2分子産生され(③)、核酸合成の材料になるリボース5リン酸が産生される(④)。がん細胞ではグルコースの取込みが増え、解糖系とペントースリン酸経路が亢進して、細胞分裂のためのエネルギー(ATP)と物質合成(核酸、脂肪酸、NADPHなど)が亢進している。2-デオキシ-D-グルコース(2-DG)は、ヘキソキナーゼ(HK)で代謝されて2-DG-6リン酸(2-DG-6-PO4)に変換され(⑤)、2-DG-6-PO4はヘキソキナーゼ(HK)とホスホグルコースイソメラーゼ(PGI)を阻害する(⑥)ので、グルコースのペントース・リン酸経路での代謝を阻害する。
【糖タンパク質の糖鎖はヘキソサミン経路で作られる】
タンパク質は遺伝子によって決められた配列によってアミノ酸が結合して作られます。タンパク質が作られるとき、まず遺伝子(DNA)からメッセンジャーRNAが転写されます。このメッセンジャーRNAからタンパク質が合成される過程を「翻訳」と言います。メッセンジャーRNAからポリペプチドへの翻訳はリボソームで行われます。
翻訳後のポリペプチド鎖は小胞体で3次元的に折り畳まれます。
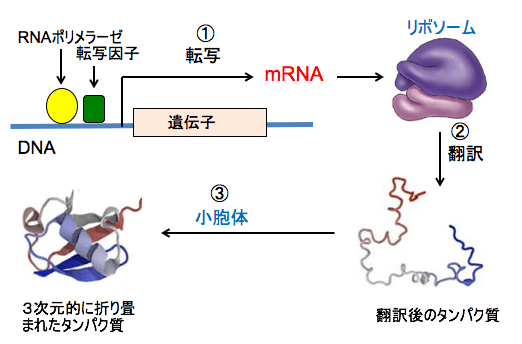
図:DNA上の遺伝子からRNAポリメラーゼや転写因子の働きによってmRNAが生成される過程を転写という(①)。mRNAの情報に基づき、リボソームにおいてアミノ酸が順番に結合してタンパク質が生成されることを翻訳という(②)。翻訳後のポリペプチド鎖は小胞体で3次元的に折り畳まれる(③)。
できたタンパク質はさらにリン酸やアセチル基や糖鎖などが結合して、タンパク質の活性や働きが変化します。このようなタンパク質の修飾を翻訳後修飾と言います(下図)。このような翻訳後修飾によってタンパク質の働きが制御されています。
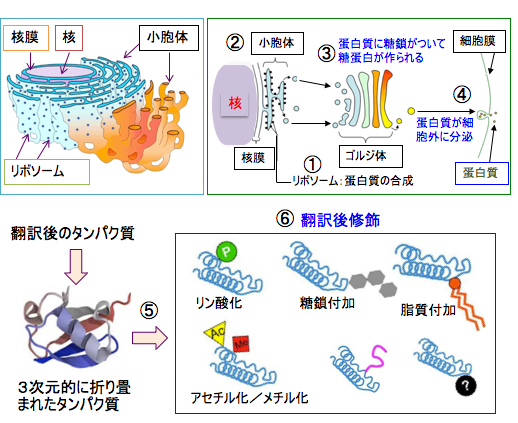
図:タンパク質はリボソームで合成され(①)、小胞体で折り畳まれ(②)、ゴルジ体で糖鎖が結合して糖タンパク質になって、細胞内や細胞外に分布して機能を発揮する(④)。翻訳後のタンパク質の多くのタンパク質はさらに、リン酸化、糖鎖付加、脂質付加、アセチル化、メチル化などの翻訳後修飾(⑥)を受けることによって機能を持つようになる。
糖鎖には、O-結合型糖鎖(セリン・スレオニン結合型糖鎖)と、N-結合型糖鎖(アスパラギン結合型糖鎖)とが存在します。
O-結合型糖鎖はアミノ酸のセリン(Ser)やスレオニン(Thr)側鎖の水酸基に結合していて、N型糖鎖はアスパラギン(Asn)残基に結合しています。
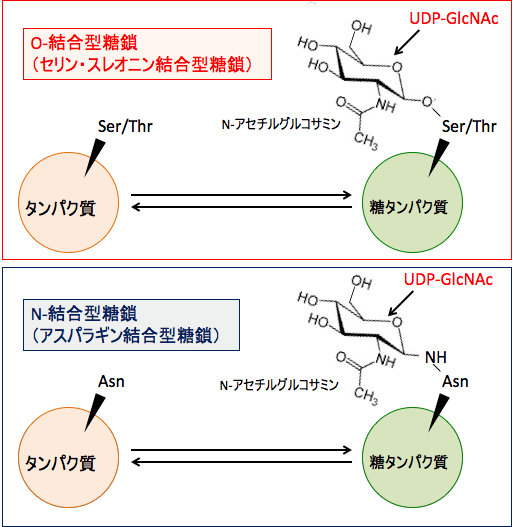
図:O-結合型糖鎖はタンパク質のセリン(Ser)やスレオニン(Thr)残基に、糖供与体のウリジン2リン酸-N-アセチルグルコサミン (UDP-GlcNAc) からのN-アセチルグルコサミンが結合している。O-結合型糖鎖では、アスパラギン(Asn)残基にN-アセチルグルコサミンが結合している。
この糖鎖結合の過程に使われるのがUDP-N-アセチルグルコサミン(UDP-GlcNAc)です。
UDP-GlcNAcは糖タンパク質、糖脂質、プロテオグリカンの合成に使われます。
このUDP-GlcNAcはヘキソサミン経路で合成されます。
解糖系のグルコース-6-リン酸の次のフルクトース-6-リン酸はヘキソサミン生合成系に入り、UDP-N-アセチルグルコサミン(UDP-GlcNAc)を産生します。
グルタミンとフルクトース6リン酸を基質として、グルタミン-フルクトース-6-リン酸アミノトランスフェラーゼ(GFAT)の働きで、グルコサミン6-リン酸が生成され、N-アセチルグルコサミン6-リン酸はN-アセチルグルコサミン1-リン酸に変換されます。
ヘキソサミン経路の最終段階では、UDP-N-アセチルグルコサミンピロホスホリラーゼ(UAP)の働きで、N-アセチルグルコサミン1-リン酸にウリジン2リン酸(UDP)が付加され、UDP-N-アセチルグルコサミン(UDP-GlcNAc)が生成します。
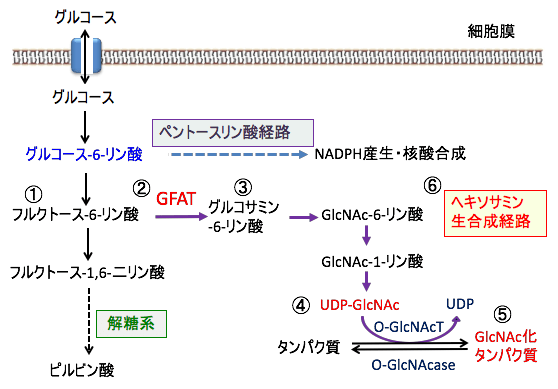
図:解糖系のフルクトース-6-リン酸(①)にグルタミン-フルクトース-6-リン酸アミノトランスフェラーゼ(GFAT)の働きで(②)、グルタミンと結合して、グルコサミン6-リン酸が生成され(③)、UDP-N-アセチルグルコサミン(UDP-GlcNAc)が生成する(④)。UDP-GlcNAcは糖タンパク質の産生に使われる(⑤)。UDP-GlcNAcを生成する経路をヘキソサミン生合成経路という(⑥)。
UDP-GlcNAcはUDPガラクトース-4-エピメラーゼによってUDP-N-アセチルガラクトサミン(UDP-GalNAc)との間で相互変換できます。
タンパク質のセリンやスレオニン残基に、糖供与体のウリジン2リン酸-N-アセチルグルコサミン (UDP-GlcNAc) からのN-アセチルグルコサミンの転移を触媒するのが、O-GlcNAc転移酵素 (OGT)で、タンパク質からN-アセチルグルコサミンを除去する酵素がO-GlcNAcase(N-アセチルグルコサミニダーゼ)です。
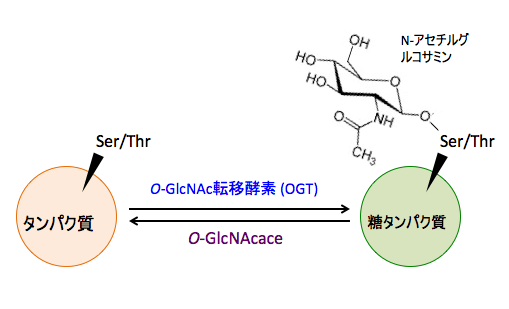
図:タンパク質のセリン(Ser)やスレオニン(Thr)にN-アセチルグルコサミンが結合することによってタンパク質の働きが制御されている。N-アセチルグルコサミンの結合は、結合を触媒する酵素(O-GlcNAc転移酵素)と除去する酵素(O-GlcNAcase)によって制御されている。
タンパク質に結合したN-アセチルグルコサミンにさらに様々な単糖が結合して糖鎖が作られます。糖鎖が結合したタンパク質を糖タンパク質と言います。多くの膜結合タンパク質や分泌タンパク質が糖タンパク質です。
上述のように、糖質とタンパク質はN-グリコシド結合もしくはO-グリコシド結合によって結合します。
この修飾は小胞体とゴルジ体で起こります。
糖タンパク質に含まれる糖質のうち主要なものは、グルコース、ガラクトース、フコース、マンノース、N-アセチルノイラミン酸、N-アセチルガラクトサミン(GalNAc)、N-アセチルグルコサミン(GlcNAc)などです。
糖鎖がタンパク質と共有結合して、たんぱく質の活性や安定性を変化させます。つまり、糖タンパク質の生成は糖代謝とシグナル伝達の接点となります。
転写因子やRNAポリメラーゼIIのようないくつかの細胞内タンパク質はO-GlcNAcの結合によって修飾されます。
【2−デオキシ-D-グルコースはヘキソサミン生合成を阻害する】
もし、正常細胞には取り込まれず、がん細胞に多く取り込まれて、ヘキソサミン生合成を阻害する物質があれば、それはがん治療に効果が期待できます。
2−デオキシ-D-グルコースがヘキソサミン生合成を阻害することが報告されています。以下のような報告があります。
2-Deoxy-d-glucose increases GFAT1 phosphorylation resulting in endoplasmic reticulum-related apoptosis via disruption of protein N-glycosylation in pancreatic cancer cells.(2-デオキシ-d-グルコースはGFAT1リン酸化を増加させ、膵臓がん細胞のタンパク質N-グリコシル化の阻害を介して小胞体関連アポトーシスを引き起こす)Biochem Biophys Res Commun. 2018 Jun 27;501(3):668-673.
【要旨】
解糖系阻害剤の2-デオキシ-d-グルコース(2DG)はエネルギー飢餓を引き起こし、多くの種類のがん細胞株の細胞生存率に影響を与える。膵臓がん細胞における2DGの作用を検討するために、2DG投与後の膵臓がん細胞株のプロテオーム解析を実施した。
2DG投与によって80個のタンパク質に発現の変化が見られた。これらの中で、ホスホヘキソース代謝に関与するタンパク質の発現亢進が認められた。
糖タンパク質を維持するために必要なウリジン二リン酸N-アセチルグルコサミン(UDP-GlcNAc)を生成するヘキソサミン生合成経路で作用するフルクトース6-リン酸アミノトランスフェラーゼ1(GFAT1)が、mRNAおよびタンパク質レベルで発現の亢進を認めた。
そこで、N糖タンパク質の総量を測定した。
予想外に、総N糖タンパク質の減少と、AMP活性化プロテインキナーゼ(AMPK)によるGFAT1のリン酸化を認めた。
これらの実験結果は、ヘキソサミン生合成経路の異常を示唆している。
さらに、2DGを投与した細胞では、グルコース応答タンパク質78(glucose response protein 78:GRP78)やC / EBP相同タンパク質(C/EBP-homologous protein :CHOP)などの小胞体ストレスマーカーの発現の増加を認め、2DGが小胞体ストレスを誘導することが示された。
さらに、メトホルミンによるAMPKの相加的活性化は、2DGの存在下でタンパク質のNグリコシル化の減少と細胞増殖阻害を相乗的に増強した。
これらの結果は、膵臓がん細胞において2DGがGFAT1のリン酸化を亢進し、タンパク質のNグリコシル化を低下させ、小胞体ストレスを亢進して細胞増殖の阻害を引き起こすことを示唆している。
つまり、2-デオキシ-D-グルコースは、ヘキソサミン生合成経路で作用するフルクトース6-リン酸アミノトランスフェラーゼ1(GFAT1)をリン酸化してタンパク質のN-グリコシル化を阻害し、小胞体ストレスを引き起こしてアポトーシスを誘導するということです。
さらにメトホルミンはAMPKを活性化して、GFAT1のリン酸化を相乗的に増やすので、2DGとメトホルミンの併用は、タンパク質のN-グリコシル化の阻害と小胞体ストレス誘導において相乗効果が期待できると言う結果です。
2-デオキシ-D-グルコースとメトホルミンの相乗的な抗がん作用については、他にも多くの報告があります。
がん治療において、2-デオキシ-D-グルコースとメトホルミンの併用は試してみる価値があります。
【2-デオキシ-D-グルコースはがん細胞に小胞体ストレスを引き起こす】
前述のように2-DGはタンパク質のN-グリコシル化(N-glycosylation)を阻害します。
糖鎖異常を起こしたタンパク質は正常な折り畳み(3次構造)ができません。
小胞体(Endoplasmic reticulum)は、細胞内における分泌・膜タンパク質の品質管理において大切な小器官です。
折り畳みに失敗した異常なタンパク質は小胞体にとどまります。
このような正常な高次構造に折り畳まれなかった異常タンパク質が小胞体内に蓄積して、細胞への悪影響(=ストレス)が生じることを小胞体ストレス(ERストレス:Endoplasmic reticulum stress)と言います。
変性タンパク質が過剰に蓄積し、小胞体ストレスの強さが細胞の回避機能を越えると、細胞死(アポトーシス)が誘導されます。小胞体ストレスはアルツハイマー病などの神経変性疾患などさまざまな疾患の原因となると考えられています。
小胞体ストレスが生じると、細胞は小胞体ストレスを軽減する応答が発動します。これを小胞体ストレス応答と言います。
小胞体に異常タンパク質が増えると、まず、タンパク質の翻訳(合成)を抑制します。さらに、折り畳み不全の異常タンパク質を正常化する分子シャペロンのGRP78と言うタンパク質の合成を亢進し、異常タンパク質の修復を行います。それでも異常タンパク質が減らなければ、異常タンパク質をプロテアソームで分解します。
しかし、小胞体ストレスが強度で長期に及んだり、小胞体ストレス応答が阻害されたりすると、細胞はアポトーシスのシグナルのスイッチが入り、自滅します。
2-DGは解糖系を阻害する以外に、タンパク質に糖鎖が着くN-グリコシル化の過程を阻害しれます。
糖鎖異常の糖タンパク質は、折り畳みが不完全な異常タンパク質になり、小胞体に蓄積して小胞体ストレスを引き起こします。
2-デオキシ-D-グルコース(2-DG)は小胞体ストレスを高めて免疫原性細胞死を増強する作用が知られています。
【小胞体ストレスを引き起こす薬物+抗がん剤=免疫原性細胞死】
抗がんや放射線を使ったがん治療の場合は、ダメージ関連分子パターン(DAMPs)を誘導する細胞死のメカニズムを利用すると、がん特異免疫を増強できることが知られています。
すなわち、DAMPsを誘導しやすくする薬剤は抗がん剤や放射線治療の抗腫瘍効果を高めることができます。
2-デオキシ-D-グルコース(2-DG)は小胞体ストレスを高めてがん細胞を死滅させる作用があります。さらに、2−デオキシ-D-グルコースは抗がん剤や放射線の免疫原性細胞死を増強することが報告されています。
2-DGとエトポシドの併用で、免疫原性細胞死を誘導し、樹状細胞が活性化され、CD8+の細胞障害性T細胞の活性が亢進することが報告されています。
抗がん剤でがん細胞を死滅させるときに2−DGを投与しておくと、死滅したがん細胞は免疫原性が高くなるので、がん抗原特異的な抗腫瘍免疫を誘導でき、延命効果を高めることができるというメカニズムです。

図:2-デオキシ-D-グルコース(2-DG)はグルコースの2位のOHがHに変わっているグルコース類縁物質で、グルコースと同様にグルコーストランスポーター(GLUT1)によって細胞内に取り込まれる(①)。細胞内のヘキソキナーゼで2-DG-6リン酸になるが、それから先の解糖系には進めない(②)。さらに2-DG-6リン酸はヘキソキナーゼを阻害するので、解糖系でのATP産生を阻害する(③)。2-DGは小胞体でのタンパク質のN-グリコシル化(糖鎖の結合による修飾)を阻害し、小胞体ストレスを引き起こす(④)。この状態で、抗がん剤や放射線でがん細胞にダメージを与えると(⑤)、小胞体のカルレチキュリンというタンパク質が死滅したがん細胞の細胞膜上に移行して「ダメージ関連分子パターン(danger-associated molecular patterns:DAMP)」として免疫細胞に認識され(⑥)、抗腫瘍免疫が活性化される(⑦)。
2-DGと抗がん剤(シクロフォスファミドやエトポシドやアントラサイクリン系)の併用投与による抗腫瘍免疫応答を活性化する治療と、抗原提示細胞やCTLの活性を増強する治療を交互に、あるいは併用して行えば、免疫系による腫瘍細胞の排除ができる可能性が高くなります。
2-DGと抗がん剤を併用すると、小胞体のカルレチキュリンというタンパク質が細胞膜上に露出して免疫原性を高めるという結果が報告されています。以下のような報告があります。
Combination of glycolysis inhibition with chemotherapy results in an antitumor immune response.(抗がん剤治療に解糖系阻害を併用すると抗腫瘍免疫応答が引き起こされる)PNAS 109 (49): 20071-20076, 2012年
【要旨】
細胞のDNAにダメージを与える抗がん剤の多くは、抗腫瘍免疫を誘導する作用がある。解糖系の亢進はがん細胞の最も良く知られた特徴の一つである。そこで、解糖系を阻害する2-デオキシグルコース(2-DG)と細胞傷害性の抗がん剤を併用した場合、抗腫瘍免疫の誘導にどのような影響を及ぼすかを検討した。
2-DGと抗がん剤のエトポシドは、免疫機能の正常なマウスにおいては相乗的に作用して寿命を延長した。しかし、免疫機能不全のマウスに対しては寿命延長効果は認められなかった。
2-DGとエトポシドの両方を投与したマウスにおいてのみ、がん細胞特異的なT細胞の十分な活性化が認められた。
さらに、2-DGとエトポシドの両方の処理によって死滅したがん細胞をマウスに免疫すると、同じ腫瘍の再度の移植に対して拒絶した。
この効果の少なくとも一部は、細胞膜上のERp57/calreticulinの出現が関連していた。
これらの結果は、がん細胞の解糖系をターゲットにすると、死滅がん細胞による通常の免疫寛容誘発性の刺激を、腫瘍免疫誘発性の刺激に変換できることを示している。このメカニズムを利用すると免疫化学療法の新しい戦略を作りだすことができる。
小胞体(Endoplasmic reticulum)は、細胞内における分泌・膜タンパク質の品質管理において大切な小器官です。
カルレチキュリンは、小胞体内腔における主要なカルシウム結合(蓄積)タンパク質として機能する多機能タンパク質です。分子シャペロンとして分泌タンパク質の品質管理の働きも行っています。また核では転写調節の働きを行っています。
2-DGは解糖系を阻害する以外に、タンパク質に糖鎖が着くN-グリコシル化の過程を阻害するので、糖タンパク質の生成を阻害します。
グリコシル化というのはタンパク質に糖類が付加する反応で、小胞体で行われて、正常に糖が付加したタンパク質はゴルジ体に運ばれます。
糖鎖異常の糖タンパク質は小胞体に蓄積して小胞体ストレスを引き起こし、細胞死の原因にもなります。(小胞体ストレスについては298話参照)
2-DGの場合は、糖タンパク質のグリコシル化が阻害され、小胞体ストレスが起こり、その状態で死滅すると死滅した細胞の細胞膜の表面にカルチキュリンが移行してダメージ関連分子パターンとなり、免疫細胞を活性化する結果、抗腫瘍免疫が活性化されるということです。
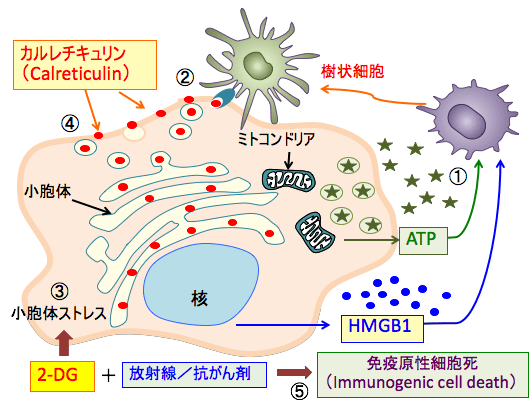
図:細胞がダメージを受けて死滅するとき、細胞内に存在する成分が放出されて炎症細胞や免疫細胞を刺激する。ミトコンドリアのATPや核のHMGB1(High-mobility group box 1 protein)は細胞外に放出されると樹状細胞を刺激する(①)。小胞体のカルレチキュリン(Calreticulin)は細胞表面に出て、樹状細胞に認識され、貪食のシグナルとなり、がん抗原を提示する働きを活性化する(②)。 2-DG(2-デオキシ-D-グルコース)は、タンパク質に糖鎖が着くN-グリコシル化の過程を阻害するので、糖鎖異常の糖タンパク質が小胞体に蓄積して小胞体ストレスを引き起こす(③)。小胞体ストレスの高い状態で放射線や抗がん剤でがん細胞が死滅するとカルレチキュリンが多く露出した死細胞となる(④)。このような免疫応答を引き起こしやすい細胞死を「免疫原性細胞死」という(⑤)。放射線治療や抗がん剤治療に2-DGを使用すると免疫原性細胞死を誘導してがん抗原に特異的な抗腫瘍免疫を高めることができる。
2DGがT細胞の抗腫瘍活性を増強することも最近明らかになっています。以下のような報告があります。
Blockade of N-Glycosylation Promotes Antitumor Immune Response of T Cells(N-グリコシル化の阻害はT細胞の抗腫瘍免疫応答を促進する)J Immunol March 1, 2020, 204 (5) 1373-1385;
この研究では、2-DGはT細胞にNK(ナチュラルキラー)細胞と類似した性質を獲得させ、がん細胞に対する殺細胞活性を高めることを報告しています。また、galectin-3によるT細胞のアポトーシス誘導を、2-DGは抑制する(T細胞がアポトーシスに抵抗性になる)ことも明らかにしています。そして、このような2-DGの作用はN-グリコシル化の阻害が関与していると報告しています。
2-デオキシ-D-グルコースはがん細胞に多く取り込まれ、解糖系を阻害します。さらに、がん細胞に作用して免疫原性細胞死を誘導するだけでなく、T細胞の免疫応答を増強することを示唆しています。がんの免疫治療において2-デオキシ-D-グルコースは試してみる価値はあると思います。
| « 692)免疫療法... | 694)免疫原性... » |