がんの予防や治療における漢方治療の存在意義を考察しています。がん治療に役立つ情報も紹介しています。
「漢方がん治療」を考える
カレンダー
| 2025年1月 | ||||||||
| 日 | 月 | 火 | 水 | 木 | 金 | 土 | ||
| 1 | 2 | 3 | 4 | |||||
| 5 | 6 | 7 | 8 | 9 | 10 | 11 | ||
| 12 | 13 | 14 | 15 | 16 | 17 | 18 | ||
| 19 | 20 | 21 | 22 | 23 | 24 | 25 | ||
| 26 | 27 | 28 | 29 | 30 | 31 | |||
|
||||||||
goo ブログ
過去の記事
カテゴリ
最新の投稿
最新のコメント
最新のトラックバック
ブックマーク
プロフィール
検索
gooおすすめリンク



| URLをメールで送信する | |
| (for PC & MOBILE) | |
834)抗腫瘍免疫増強法(その3):再利用薬を用いた免疫チェックポイント阻害
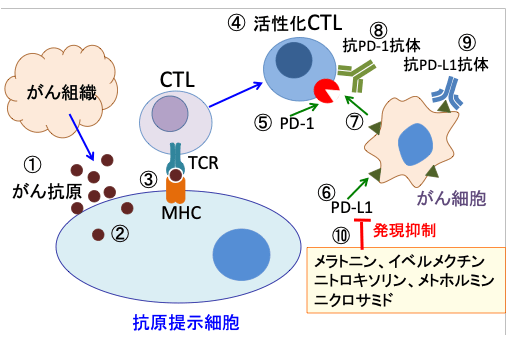
図:がん細胞から放出されたがん抗原(①)は、樹状細胞やマクロファージなどの抗原提示細胞に取込まれ(②)、ペプチドに分解されて抗原ペプチドとして抗原提示細胞上のMHC(major histocompatibility complex:主要組織適合抗原複合体)に提示される(③)。MHCはがん抗原を介してCTL(細胞傷害性T細胞)上のTCR(T細胞受容体)と反応してCTLを活性化する(④)。CTLは活性化されるとPD-1(Programmed death-1)が発現する(⑤)。がん細胞にはPD-1のリガンドであるPD-L1が発現している(⑥)。PD-1とPD-L1が結合するとCTLは増殖が抑制される(⑦)。PD-1とPD-L1の結合を抗PD-1抗体(⑧)や抗PD-L1抗体(⑨)で阻止するとCTLの抗腫瘍活性を高めることができる。メラトニン、イベルメクチン、ニトロキソリン、メトホルミン、ニクロサミドはがん細胞のPD-L1の発現量を減らし、細胞傷害性T細胞(CTL)による抗腫瘍効果を高めることができる(⑩)。
834)抗腫瘍免疫増強法(その3):再利用薬を用いた免疫チェックポイント阻害
【細胞傷害性T細胞を抑制するPD-1とCTLA-4】
細胞傷害性T細胞(キラーT細胞)は抗原提示細胞(樹状細胞やマクロファージ)から抗原を提示されると活性化されて、敵(病原菌やがん細胞など)を攻撃します。
細胞傷害性T細胞にはPD-1やCTLA-4という受容体が存在します。PD-1はプログラム細胞死1(programmed death-1)、CTLA-4は細胞傷害性Tリンパ球抗原-4 (cytotoxic T-lymphocyte-associated protein 4)の略です。
これらの受容体のリガンド(受容体に結合して作用する物質)となるPD-L1やB7(B7-1, B7-2)を抗原提示細胞が持つことによって細胞傷害性T細胞の働きを抑制しています。
つまり、PD-1受容体やCTLA-4受容体がリガンドによって刺激されると、T細胞の増殖が停止し細胞死を来すことになります。このようにして細胞傷害性T細胞の過剰な応答を制御しています。
細胞傷害性T細胞の働きを阻害するPD-L1やB7はがん細胞にも発現しています。つまり、がん細胞は免疫系の制御システムを利用して、がん組織内の細胞傷害性T細胞の働きを阻止しています。
PD-1受容体やCTLA-4受容体は細胞傷害性T細胞を死滅させるスイッチなようなものなので、これらのスイッチが入らないようにすれば、細胞傷害性T細胞は生き残ってがん細胞の攻撃力を高めることができます。
CTLA-4に対する抗体(ヒト型抗ヒトCTLA-4モノクローナル抗体)のイピリブマブ(ipilimumab: YERVOY)やヒト型抗PD-1モノクローナル抗体のニボルマブ(nivolumab商品名「オプジーボ(Opdivo)」)などがあります。このような免疫チェックポイント阻害剤を使用すると、がん細胞を攻撃する細胞傷害性T細胞の働きを高めることが可能になります。
体に備わったがん細胞に対する攻撃力を高めてがんを治療しようというのが「がんの免疫療法」の理論です。「免疫細胞を活性化する」という従来の免疫療法では十分な効果が得られなかったのですが、その大きな理由は免疫応答にブレーキをかける仕組みの存在です。このブレーキを解除して免疫細胞に100%の力でがん細胞を攻撃させようというのが、CTLA-4やPD-1/PD-L1をターゲットにした治療法です。(下図)
ただ、この治療法は免疫細胞の暴走を許して、自己免疫疾患を引き起こすという副作用もあります。

図:抗原提示細胞上にはMHCクラスII(MHC-II)といわれる分子があり、抗原を介してT細胞上のTCR(T細胞受容体)と反応して細胞傷害性T細胞を活性化する(①)。T細胞上にはCD28とCTLA-4があり、CD28は恒常的に発現し、抗原提示細胞からのB7-1やB7-2というリガンドによってT細胞活性化に作用する(②)。一方、CTLA-4はT細胞活性化にともなって発現が誘導され、B7-1やB7-2によって刺激されるとT細胞を抑制する(③)。CTLA-4はCD28よりもB7に対する親和性が強いので、活性化したT細胞の過剰な応答を抑制する。同様に、PD-1(Programmed death-1)は抗原提示細胞のPD-L1(別名B7-H1)と結合することによって抑制型の免疫調節シグナルを活性化させる(④)。がん細胞もB7-1やB7-2やPD-L1が発現しており、細胞傷害性T細胞の働きを抑制している。T細胞のCTLA-4とPD-1の働きを特異抗体で阻害すると、がん細胞に対する細胞傷害性T細胞の働きを高めることができる(⑤)。
がん細胞を非自己と認識して、それを攻撃するためにT細胞は活性化しますが、PD-1リガンド(PD-L1)を持ったがん細胞と接触すると、CTL上のPD-1とリガンド(PD-L1)が結合することにより、免疫シグナルは抑制され、T細胞はがん細胞を攻撃できなくなってしまいます。これがT細胞を活性化するだけの従来の免疫療法に限界があった理由です。活性化したCTL(細胞傷害性T細胞)をがん組織に送っても、がん細胞を攻撃しようと近づくとPD-L1によって自身のPD-1のスイッチが入って死滅するからです。
がん細胞がPD-1リガンド(PD-L1)を多く発現しているほど、予後が悪いというデータも報告されています。
T細胞やNK細胞を活性化すると同時に、T細胞上のPD-1やCTLA-4の働きを阻止すると、がん組織を免疫力だけで縮小できる可能性が高まります。
【がん細胞は低酸素誘導因子-1(HIF-1)の発現が亢進している】
がん組織は酸素の供給が不足し、低酸素状態になっています。低酸素は低酸素誘導因子-1(Hypoxia Inducible Factor-1: HIF-1)という転写因子を活性化します。このHIF-1は何百という遺伝子の発現を亢進し、細胞は低酸素に適応できるようになります。
がん細胞の特徴的な代謝異常であるワールブルグ効果(解答系と乳酸産生の亢進と、ミトコンドリアでの酸化的リン酸化の抑制)を根本で制御しているのがHIF-1です。
HIF-1はグルコースを取り込むGLUT-1の発現を亢進し、解糖系酵素の発現を亢進します。一方、ピルビン酸脱水素酵素キナーゼの発現を亢進してピルビン酸脱水素酵素の活性を阻害し、ミトコンドリアの酸化的リン酸化を抑制します。つまり、HIF-1の活性亢進がワールブルグ効果を引き起こしていると言えます。

図:酸素分圧(pO2)が低下して低酸素になると(①)、低酸素誘導因子-1(HIF-1)の発現が亢進する(②)。HIF-1はグルコースを取り込むGLUT-1(③)と解糖系酵素(④)と乳酸を排出するMCT4(⑤)の発現を亢進する。HIF-1は血管内皮細胞増殖因子(VEGF)の産生を増やして血管新生を亢進する(⑥)。ペントース・リン酸経路を亢進し(⑦)、NADPHと核酸の合成を促進する(⑧)。HIF-1はピルビン酸脱水素酵素キナーゼの発現を亢進し(⑨)、ピルビン酸脱水素酵素の活性を阻害し、アセチルCoAの産生を低下させ、ミトコンドリアでの代謝を抑制する(⑩)。つまり、HIF-1は解糖系を亢進し、ミトコンドリアの酸化的リン酸化を抑制してワールブルグ効果を促進する。
がん細胞の代謝の特徴は、酸素が十分に利用できる状況でも、酸素を使わない解糖系が亢進し、ミトコンドリアでの酸素を使ったエネルギー産生(酸化的リン酸化)が抑制されていることです。
このような代謝の特徴の根本的なメカニズムは、がん細胞では酸素濃度とは関係なく、恒常的にHIF-1が活性化しているためです。つまり、がん細胞では恒常的に低酸素シグナルがオンになっているということです。その理由は、がん細胞で活性化されているmTORC1やSTAT3がHIF-1の産生を促進するからです。
がん細胞の増殖シグナル伝達系であるPI-3キナーゼ/Akt/mTORC1シグナル伝達系においてmTORC1はHIF-1のタンパク質の産生(mRNAからタンパク質の翻訳)を促進します。また、増殖因子やサイトカインで活性化されるSTAT3という転写因子はHIF-1遺伝子の転写を亢進します。
mTORC1(哺乳類ラパマイシン標的タンパク質複合体1)はリボソームの生合成を促進するS6Kをリン酸化して活性化する作用によってタンパク質合成を促進し、HIF-1タンパク質の産生を増やします。
一方、STAT(signal transducer and activator of transcription;シグナル伝達兼転写活性化因子)は、様々な増殖因子やサイトカインを中心とする細胞外からの刺激によって活性化されたJAKなどのチロシンキナーゼによってリン酸化を受けると2量体を形成し、核内に移行してさまざまな遺伝子の発現を誘導します。
がん細胞におけるmTORC1とSTAT3の恒常的な活性化が、低酸素が無くてもHIF-1の産生を促進するのです。

図:増殖刺激や遺伝子変異などによってがん細胞で恒常的に活性が亢進しているSTAT3(シグナル伝達兼転写活性化因子)は低酸素誘導因子-1(HIF-1)遺伝子の転写(mRNAの産生)を促進し、mTORC1はリボソームの生合成を促進するS6Kを活性化してHIF-1タンパク質の合成を促進する。その結果、がん細胞では低酸素でなくてもHIF-1タンパク質の合成が亢進している。
【低酸素誘導因子-1(HIF-1)はPD-L1の発現を亢進する】
がん組織で発現と活性が亢進している低酸素誘導因子-1(HIF-1)はPD-L1の発現を亢進することが多くの研究で明らかになっています。例えば以下のような報告があります。
PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation(PD-L1はHIF-1αの新しい直接標的であり、低酸素下でのその遮断は骨髄由来抑制細胞を介したT細胞活性化を促進する)J Exp Med. 2014 May 5;211(5):781-90.
【要旨の抜粋】
骨髄由来抑制細胞において、免疫チェックポイント受容体 (PD-1および CTLA-4) とそれぞれのリガンド (PD-L1、PD-L2、CD80、および CD86) に対する低酸素症の影響を調査した。
低酸素症は、担がんマウスの脾臓の骨髄由来抑制細胞で PD-L1 の急速かつ劇的かつ選択的な発現亢進を引き起こした。この現象は骨髄由来抑制細胞に限定されたものではなく、マクロファージ、樹状細胞、および腫瘍細胞でも低酸素はPD-L1の発現を有意に増加させた。
さらに、低酸素下での PD-L1 発現亢進は、低酸素誘導因子-1α (HIF-1α) に依存していた。HIF-1α が PD-L1 遺伝子の転写プロモーター領域の低酸素応答エレメントに直接結合することが明らかになった。
HIF-1αの阻害と同時にPD-L1を遮断することは、がん免疫療法の新しいアプローチとなる可能性がある。
低酸素誘導因子-1(HIF-1)が骨髄由来抑制細胞やマクロファージ、樹状細胞、がん細胞におけるPD-L1の発現を亢進するという報告です。免疫チェックポイント阻害剤を使うとき、HIF-1の活性を阻害する方法の併用の有効性を示唆しています。
以下のような報告もあります。
HIF inhibitor 32-134D eradicates murine hepatocellular carcinoma in combination with anti-PD1 therapy(HIF 阻害剤 32-134D は抗 PD1 療法と組み合わせてマウス肝細胞癌を根絶する)J Clin Invest. 2022 May 2;132(9):e156774.
肝臓がん細胞を移植したマウスを用いた動物実験で、抗 PD1 療法に開発中の低酸素誘導因子(HIF)阻害剤の32-134Dを併用すると腫瘍根絶率が 25% から 67% に増加しました。
がん細胞の免疫回避を媒介する腫瘍関連マクロファージおよび骨髄由来抑制細胞が減少し、がん細胞を攻撃する CD8+ T 細胞およびナチュラルキラー細胞の増加が認められました。
免疫チェックポイント阻害剤の治療に低酸素誘導因子-1(HIF-1)の阻害剤を併用すると、抗腫瘍効果を高めることができることを示しています。
【メラトニンは低酸素誘導因子-1の活性化を阻止する】
生物は外界の酸素濃度を認識する巧みな仕組みを保持しています。
酸素濃度が低下すると、生物は低酸素シグナルを活性化し低酸素状態に適応します。この低酸素応答の中心的分子が低酸素誘導因子-1(Hypoxia inducible factor-1: HIF-1) およびプロリル・ヒドロキシラーゼ(prolyl hydroxylase)と呼ばれる2つのタンパク質です。
HIF-1は、細胞が酸素不足に陥った際に誘導されてくる転写因子です。αとβの2つのサブユニットからなるヘテロ二量体であり、βサブユニットは定常的に発現して細胞核にいますが、 HIF-1αは細胞質で酸素濃度依存的な分解を受けます。
HIF-1αは、正常酸素濃度下では、HIF-1αタンパク質中の2カ所のプロリン残基がプロリル・ヒドロキシラーゼにより水酸化されることによりVHL(von Hippel-Lindau)タンパク質が結合します。
VHLが結合するとHIF-1αのユビキチン化が促進されて26Sプロテアソームで分解されます。したがって、酸素が十分にある状況ではHIF-1は不活性の状態に維持されます。
プロリル・ヒドロキシラーゼは酸素濃度感受性のタンパク質で、酸素濃度が低下するとプロリル・ヒドロキシラーゼの酵素活性が著しく低下します。すると、HIF-1αのプロリン残基の水酸化が起きないので、HIF-1αは分解を受けずに安定化します。
安定化したHIF-1αは核内に移行し、HIF-1βと二量体を形成して低酸素応答配列(Hypoxia Responsive Element)に結合して、低酸素応答に必要な様々な遺伝子の発現を活性化します。
HIF-1は各種解糖系酵素、グルコース輸送蛋白、血管内皮増殖因子(VEGF)、造血因子エリスロポイエチンなど多くの遺伝子の発現を転写レベルで制御し、細胞から組織・個体にいたる全てのレベルの低酸素適応反応を制御しています(下図)。

図:酸素濃度が高い状態では、HIF-1αは酸素濃度感受性タンパク質のプロリル・ヒドロキシラーゼによって水酸化され(①)、VHL(von Hippel-Lindau)タンパク質が結合して26Sプロテアソームで分解される(②)。低酸素状態ではプロリル・ヒドロキシラーゼの活性が低下してHIF-1αの分解が阻止されるので、蓄積したHIF-1αは核内に移行し(③)、HIF-1βとヘテロダイマー(ヘテロ二量体)を形成して遺伝子の低酸素応答配列に結合し(④)、コアクチベーター(CBP/p300)やRNAポリメラーゼがリクルートされて遺伝子転写を亢進し(⑤)、低酸素状態の適応に必要な様々な遺伝子の発現を誘導する(⑥)。
メラトニンがHIF-1αの分解を促進してHIF-1αタンパク量を減らすことが報告されています。
このHIF-1αを抑制するメラトニンの作用は抗酸化作用が関連しています。抗酸化作用によって、メラトニンはHIF-1αタンパク質の分解を促進します。
メラトニンはフリーラジカルを消去することによって、活性酸素によるプロリル・ヒドロキシラーゼの不活性化(酵素の二価鉄イオンを酸化することによって)を阻止し、HIF-1αの分解を亢進してHIF-1αの量を減らします。すなわち、メラトニンはHIF-1αのmRNA量には影響せず、HIF-1αの核への移行を阻止し、HIF-1αとそのco-activatorであるCBPとp300の相互作用を阻害します。
メラトニンには免疫増強作用やがん細胞の直接的な抑制作用がありますが、がん細胞のHIF-1αの分解を促進してワールブルグ効果と血管新生を阻害する効果もメラトニンの抗腫瘍効果のメカニズムとして重要です。以下のような論文があります。
Melatonin as an angiogenesis inhibitor to combat cancer: Mechanistic evidence.(がんと戦うための血管新生阻害剤としてのメラトニン:メカニズム的証拠)Toxicol Appl Pharmacol. 2017 Nov 15;335:56-63.
この論文では、メラトニンが転写因子の低酸素誘導因子-1α(HIF-1α)の活性化を阻止し、血管内皮細胞増殖因子(VEGF)の産生を阻害して血管新生阻害作用を発揮することを報告しています。
【メラトニンはPD-L1の発現を抑制する】
メラトニンが低酸素誘導因子-1(HIF-1)の発現量を減らすので、PD-L1の発現を抑制する作用も推測できます。実際に、メラトニンがPD-L1の発現を抑制することが報告されています。
以下のような報告があります。
Melatonin Downregulates PD-L1 Expression and Modulates Tumor Immunity in KRAS-Mutant Non-Small Cell Lung Cancer.(メラトニンはKRAS変異型非小細胞肺がんのPD-L1発現を低下し、腫瘍免疫を調節する)Int. J. Mol. Sci. 2021, 22(11), 5649
【要旨の抜粋】
KRAS変異を有する非小細胞肺がん患者は、化学療法や免疫療法に対して抵抗性を示す。KRAS変異非小細胞肺がんに対する免疫療法の効果を高める戦略が求められている。
本研究では、メラトニンは、A549、H460、およびLLC1細胞を含むKRAS変異の非小細胞性肺がん細胞株で細胞アポトーシスを誘導し、細胞生存率を大幅に低下させた。
KRAS変異を有する肺がん細胞は、PD-L1(programmed death ligand 1)の発現の亢進が認められた。しかし、メラトニンによる治療は、インターフェロン(IFN)-γ刺激の存在下と非存在下の両方でPD-L1発現を低下した。
さらに、KRAS変異肺がん細胞はより高いYes関連タンパク質(YAP)およびPDZ結合モチーフ(TAZ)レベルの転写共活性化因子を示し、PD-L1発現は肺がん細胞のYAPおよびTAZと正の相関が認められた。メラトニンによる治療は、YAP/TAZ下流遺伝子発現のダウンレギュレーションを伴うYAPおよびTAZを効果的に抑制した。メラトニンとYAP/TAZ阻害剤の組み合わせは、YAPとPD-L1の発現を確実に減少させた。
動物実験はさらに、メラトニンの投与が同系マウスモデルにおいて腫瘍増殖を有意に阻害し、腫瘍免疫を調節することを明らかにした。
以上から、これらのデータは、YAP / PD-L1軸を抑制することによって免疫抑制腫瘍微小環境を調節するメラトニンの新しい抗腫瘍メカニズムを明らかにし、非小細胞性肺がんを治療するためのメラトニンの治療可能性を示唆している。
YAP(Yes-associated protein)とTAZ(transcriptional co-activator with PDZ-binding motif) は転写共役因子です。
がん細胞においては、アポトーシス抵抗性、増殖促進、幹細胞の性状(stemness)に関与する遺伝子の発現を亢進するので、YAP/TAZはがん細胞の増殖を促進することになり、YAP/TAZの活性化を阻害することが治療になります。
多くのがん細胞でYAP/TAZの活性が亢進しています。変異KRASがYAPを活性化して発がんを引き起こすことが報告されています。
この論文では、メラトニンがYAP / PD-L1軸を抑制することによって免疫抑制性の腫瘍微小環境を改善する効果を示しています。
【PAK-1はPD-L1の発現を亢進する】
がん細胞で活性化しているPAK-1(p21活性化キナーゼ-1)がPD-L1の発現を亢進するので、PAK-1の阻害がPD-L1の発現を抑制して、免疫チェックポイント阻害剤などのがんの免疫療法の効き目を高めることが報告されています。
細胞の運動性を制御する細胞骨格の動的変化の背後にあるシグナル伝達経路にはRas関連の低分子量GTAアーゼ(Ras-related small GTPases)と、p21活性化キナーゼ(p21-activated kinases :PAKs)を含むエフェクタータンパク質が関与しています。
PAKキナーゼは酵母やショウジョウバエにも存在するセリン/スレオニンプロテインキナーゼのファミリーであり、哺乳類では6つのアイソフォーム(PAK1〜6)が発見されています。それらはすべて低分子量GTPaseの RacおよびCdc42の直接の標的です。
細胞骨格の動的変化の制御における役割に加えて、PAKは細胞の生存、分裂、遺伝子転写などの様々な細胞活動を調節することが明らかになっています。
いくつかの成長因子受容体チロシンキナーゼ(インスリン、IGF-1、EGF、PDGF、VEGF受容体など)およびGタンパク質共役受容体からのシグナルは、PAKの活性化につながります。
これらの経路は、PI-3キナーゼ(PI3K)とグアニンヌクレオチド交換因子(GEF)の連続的な活性化を通じて低分子量GTPase のRacおよびCdc42を活性化し、PAKを活性化します。
がん細胞では、PAKの活性化は変異Rasを介して頻繁に起こっています。
Rasは最も一般的に変異しているがん遺伝子の1つであり、MAPキナーゼ経路とPI3キナーゼを活性化し、PAKキナーゼを活性化します。活性化したPAK-1はがん細胞の増殖と転移を亢進し、PD-L1の発現を亢進して抗腫瘍免疫を抑制します。

図:受容体チロシンキナーゼ(インスリン受容体、IGF-1受容体、EGF受容体、PDGF受容体、VEGF受容体など)およびGタンパク質共役型受容体(G protein coupled receptor : GPCR)からのシグナル(①と②)は、RASおよびPI3Kを活性化し(③)、GDP/GTP交換反応を促進するGEF(guanine-nucleotide exchange factors)の作用によって(④)、Rac/Cdc42はGTP結合型になって活性化し(⑤)、Rac/Cdc42のエフェクターであるp21活性化キナーゼ(PAK-1)を活性化する(⑥)。活性化したPAK-1はがん細胞の増殖と転移を亢進し、PD-L1の発現を亢進して抗腫瘍免疫を抑制する(⑦)。さらに、PAK-1はRASによるMAPキナーゼ(分裂促進因子活性化タンパク質キナーゼ)経路の活性化を増強する(⑧)。
【イベルメクチンはPAK-1を阻害してPD-L1の発現を抑制する可能性がある】
PAK-1はPD-L1の発現を亢進します。したがって、PAK-1阻害はPD-L1の発現抑制によって抗腫瘍免疫を高めることができます。以下のような報告があります。
Inhibition of PAK1 suppresses pancreatic cancer by stimulation of anti-tumour immunity through down-regulation of PD-L1. (PAK1の阻害は、PD-L1のダウンレギュレーションによる抗腫瘍免疫の刺激により、膵臓がんを抑制する)Cancer Lett. 2020 Mar 1;472:8-18.
【要旨】
膵臓がんに対する免疫療法は、活性化した膵星細胞による結合組織の増生などによる免疫抑制性の微小環境の存在のため、優位な臨床的利益をもたらしていない。この研究は、抗腫瘍免疫におけるPAK-1の関与を調査することを目的としている。
マウスの膵臓がんのモデルでは、PAK-1遺伝子のノックアウトにより腫瘍組織内のCD4+ T細胞およびCD8+ T細胞が増加し、膵臓星細胞の活性化が阻害され、生存期間が延長した。
PAK-1の阻害は、膵星細胞の刺激による膵臓がん細胞の増殖と遊走を阻害し、細胞傷害性リンパ球による膵臓がん細胞の細胞死誘導に対する膵星細胞による保護作用を阻止し、膵臓がん細胞における内因性および星細胞刺激によるPD-L1発現を減少させ、膵臓がん細胞の細胞傷害性リンパ球による細胞死を増加した。
PAK-1の阻害は、腫瘍組織内のCD4+およびCD8+ T細胞を増加させ、内因性および星細胞誘導性のPD-L1発現のダウンレギュレーションを介して細胞傷害性リンパ球による殺傷に対して膵臓がん細胞を感作することにより、抗腫瘍免疫を刺激する。
特に免疫チェックポイント阻害剤と組み合わせたPAK-1阻害剤は、膵臓がんの免疫療法の有効性を改善する可能性がある。
PAK-1を阻害する薬としてイベルメクチン(ivermectin)があります。イベルメクチンはマクロライド類に属する物質で、腸管糞線虫症や糸状虫や疥癬など多くの寄生虫に有効です。
静岡県伊東市内のゴルフ場近くで採取した土壌から大村智博士により発見された新種の放線菌「ストレプトマイセス・アベルメクチニウス」(Streptomyces avermitilis)が生産するアベルメクチンを元に創製されました。大村智博士はこの発見で2015年にノーベル生理学・医学賞を受賞しています。
イベルメクチンがPAK-1の活性を阻害することが報告されています。
以下のような報告があります。PAK研究の第一人者の丸田浩博士たちの研究報告です。
Ivermectin inactivates the kinase PAK1 and blocks the PAK1-dependent growth of human ovarian cancer and NF2 tumor cell lines.(イベルメクチンは、キナーゼPAK1を不活性化し、ヒト卵巣がんおよびNF2腫瘍細胞株のPAK1依存性の成長を阻止する)Drug Discov Ther. 2009 Dec;3(6):243-6.
【要旨】
イベルメクチンは、線虫のGABA(γ-アミノ酪酸)受容体を阻害し、哺乳類の対応する受容体には作用しないので、線虫を非常に低用量(0.2 mg / kg)で選択的に死滅する寄生虫治療薬である。
数年前にロシアのグループから、イベルメクチンは、マウスに悪影響を与えることなく、はるかに高い用量(3-5 mg / kg)でマウスのヒトメラノーマおよび他のいくつかのがん異種移植片の成長をほぼ完全に抑制できることが報告された。しかし、その抗がん機序は分子レベルでまだ解明されていない。
イベルメクチンのPAK1阻害作用の可能性に対する最初のヒントは、致死量以下のイベルメクチンが線虫の産卵数を劇的に減少させるという最近の発見であった。
PAK1遺伝子欠損(遺伝子ノックアウト)、またはプロポリスの主要な抗がん成分であるCAPE(カフェ酸フェネチルエステル)やARC(アルテピリンC)などの天然のPAK1阻害剤による治療も、全く同じ効果を引き起こす。この事実は、キナーゼPAK1がイベルメクチンの新しい標的である可能性を示唆している。
このキナーゼは、膵臓がん、結腸がん、乳がん、前立腺がん、NF(神経線維腫症)腫瘍などのヒトのがんの70%以上の増殖に必要である。
この研究では、イベルメクチンがヒト卵巣がんおよびNF2欠損シュワン細胞腫細胞株の発がん性キナーゼPAK1を阻害し、細胞培養におけるPAK1依存性の増殖を抑制することを初めて実証した。50%増殖阻害濃度(IC50)は細胞株に応じて5〜20μMの間であった。
他にも多くの研究グループからイベルメクチンのPAK-1阻害作用が報告されています。(674話参照)
しかし、人体内でイベルメクチンのPAK-1阻害作用を期待するには、寄生虫疾患に使用する量の10倍以上の服用量が必要である可能性があります。イベルメクチンは服用法の工夫で血中濃度を高めることができます。
【メトホルミンは免疫チェックポイント阻害剤の効き目を高める】
メトホルミンはビグアナイド系薬剤に分類される経口糖尿病治療薬の一種です。様々なメカニズムで抗がん作用を発揮します。メトホルミンはAMP活性化プロテインキナーゼ(AMPK)を活性化します。AMPKの活性化はインスリン感受性を高めて血糖を低下させます。さらにAMPKの活性化はがん細胞の増殖を抑制します。
メトホルミンが免疫チェックポイント阻害剤の効き目を高める作用が報告されています。以下のような報告があります。
Metformin Promotes Antitumor Immunity via Endoplasmic-Reticulum-Associated Degradation of PD-L1.(メトホルミンはPD-L1の小胞体関連の分解を介して抗腫瘍免疫を増強する)Mol Cell. 2018 Aug 16;71(4):606-620.
【要旨】
メトホルミンは抗腫瘍活性を有し、さらに細胞傷害性Tリンパ球による免疫監視機構を増強することが報告されている。ただし、がん免疫におけるメトホルミンの役割の機能と詳細なメカニズムは完全には理解されていない。ここでは、メトホルミンがプログラム細胞死リガンド-1(programmed death ligand-1:PD-L1)の安定性と膜局在を減らすことによって細胞傷害性Tリンパ球活性を高めることを示す。
さらに、メトホルミンによって活性化されたAMP活性化プロテインキナーゼ(AMPK)が直接PD-L1のセリン195をリン酸化することを発見した。 セリン195のリン酸化は異常なPD-L1のグリコシル化を誘導し、その結果、小胞体での蓄積および小胞体関連タンパク質分解を誘導した。
この結果と一致するように、メトホルミン治療を受けた乳がん患者の腫瘍組織は、AMPK活性化を伴うPD-L1レベルの低下を示した。メトホルミンによるPD-L1の阻害シグナルの遮断はがん細胞に対する細胞傷害性Tリンパ球活性を増強する。
この実験結果は小胞体関連タンパク質分解を介するPD-L1発現の新しい調節メカニズムを明らかにし、メトホルミンと免疫チェックポイント阻害剤の組み合わせが免疫療法の有効性を高める可能性があることを示唆している。
メトホルミンはオプジーボやキイトルーダなどの免疫チェックポイント阻害剤の抗腫瘍効果を高める効果が期待できると言うことです。以下のような報告もあります。
Efficacy of metformin in combination with immune checkpoint inhibitors (anti-PD-1/anti-CTLA-4) in metastatic malignant melanoma.(転移のある悪性黒色腫における免疫チェックポイント阻害剤(抗PD-1抗体/抗CTLA-4抗体)との併用におけるメトホルミンの効果)J Immunother Cancer. 2018 Jul 2;6(1):64.
この報告は、転移性悪性黒色腫と診断され、イピリムマブ(ipilimumab)、ニボルマブ(nivolumab)および/またはペンブロリズマブ(pembrolizumab)で治療された患者を含む後ろ向きコホート研究です。免疫チョックポイント阻害剤のみで治療を受けた群と、免疫チェックポイント阻害剤にメトホルミンを併用した群で、抗腫瘍効果を比較しています。
免疫チェックポイント阻害剤とメトホルミンの併用による治療は、免疫チェックポイント阻害剤単独の治療より良好な治療関連アウトカム(客観的反応率、疾患コントロール率、無増悪生存期間および全生存期間)が観察されました。
メトホルミンが免疫チェックポイント阻害剤の効果を高める効果は、その作用メカニズムから十分に期待できると思います。
【BETファミリータンパク質ががん治療のターゲットとして注目されている】
BETファミリータンパク質は高アセチル化ヒストンへの結合を介して、がん遺伝子や抗アポトーシスタンパク質の発現を促進する作用があります。
ブロモドメインはヒストンのアセチル化リシンを認識し,制御タンパク質を集めてクロマチン構造や遺伝子発現を制御する機能が知られているタンパク質ドメインです。
ブロモドメイン繰り返し配列および特異的末端配列を持つBET(bromodomain and extra-terminal)ファミリータンパク質としてBRD2,BRD3,BRD4,BRDTが知られています。
ヒストンのアセチル化による遺伝子発現の制御には、アセチル化を促進するヒストンアセチル基転移酵素、ヒストンからアセチル基を除去するヒストン脱アセチル化酵素、ヒストンのアセチル化した部分を認識するBETファミリータンパク質の3つが必要ということです。
ヒストンアセチル基転移酵素の「書き屋(Writer)」とヒストン脱アセチル化酵素の「消し屋(Eraser)」と、BETファミリータンパク質の「読み屋(Reader)」の3つの役割を担うタンパク質が、ヒストンアセチル化の制御を行ってます(下図)。

図:「書き屋(Writer)」のヒストンアセチル基転移酵素によってヒストンにアセチル基が結合し(①)、「消し屋(Eraser)」のヒストン脱アセチル化酵素によってアセチル基が除去される(②)。ヒストンアセチル化の少ない部分では遺伝子転写は抑制される(③)。「読み屋(Reader)」のBETファミリータンパク質はヒストンのアセチル化リシンと結合して(④)、転写を制御するタンパク質をリクルートして遺伝子転写を促進する(⑤)。
BETファミリータンパク質は、c-Mycなどのがん遺伝子やBcl-2などの抗アポトーシスタンパク質の転写を亢進します。
したがって、ヒストンのアセチル化リシンとBETファミリータンパク質のブロモドメインの結合を阻害する薬剤をがん細胞に投与すると、遺伝子発現パターンが正常細胞に近づくことが知られています。このようなBET阻害剤は、ある種のがん細胞に投与すると腫瘍促進遺伝子の発現を選択的に抑制することから、がん治療薬としての可能性が期待されています。
【ニトロキソリンはBETタンパク質を阻害する】
抗生物質のニトロキソリンがBETタンパク質を阻害することが報告されています。以下のような論文があります。
Discovery of novel BET inhibitors by drug repurposing of nitroxoline and its analogues.(ニトロキソリンとその類似体の薬物再利用による新規BET阻害剤の発見)Org Biomol Chem. 2017 Nov 15;15(44):9352-9361.
薬物ライブラリーからBRD4(ブロモドメイン含有タンパク質4)特異的阻害作用を有する化合物を探索し、ニトロキソリンが、BRD4の第一ブロモドメインとアセチル化ヒストン4ペプチドとの間の相互作用を50%阻害濃度(IC50)が0.98μMで阻害することを報告しています。
ニトロキソリンは、非BETブロモドメイン含有タンパク質に対して阻害作用を示さず、良好な選択性で全てのBETファミリーメンバーを阻害しました。従ってニトロキソリンは選択的BET阻害剤と言えます。
BET (bromodomain and extra-terminal)ファミリータンパク質は、ブロモドメインにおいてヒストンのアセチル化されたリシンを認識することにより、転写活性化因子として機能します。BRD4はこのようなBETファミリータンパク質の1つです。
BETファミリータンパク質のBRD4の阻害剤(=ニトロキソリン)はc-Mycなどのがん遺伝子やBcl-2などの抗アポトーシス・タンパク質の発現を抑制する作用によって抗腫瘍効果を発揮します。

図:ヒストン脱アセチル化酵素によってヒストンのアセチル化が低下するとクロマチンが凝集して遺伝子転写活性は抑制される(①)。ヒストンアセチル基転移酵素によってヒストンがアセチル化されるとクロマチンが緩み、遺伝子転写活性が亢進する(②)。ヒストンのアセチル化されたリシンを認識するブロモドメインの繰り返し配列と特異的末端配列を持つBET(bromodomain and extra-terminal)ファミリータンパク質(③)は、ヒストンのアセチル化リシンに結合し、転写因子やRNAポリメラーゼなどの転写に必要なタンパク質をリクルートして、がん遺伝子(c-Mycなど)や抗アポトーシスタンパク質(Bcl-2など)の転写を活性化する(④)。その結果、がん細胞の増殖を促進し、細胞死(アポトーシス)に抵抗性になる(⑤)。ニトロキソリンはアセチル化リシンとブロモドメインの結合を阻害する(⑥)。その結果、がん細胞の増殖を抑制し、細胞死を誘導する。
ニトロキソリンの尿路感染症の治療に使う量は1日に500から750mgです。この尿路感染症に使用する服用量で十分な抗腫瘍効果が期待できることが動物実験の研究で報告されています。以下のような報告もあります。
Rationally repurposed nitroxoline inhibits preclinical models of Epstein-Barr virus-associated lymphoproliferation.(合理的に再利用されたニトロキソリンは、エプスタイン-バーウイルス関連リンパ球増殖の前臨床モデルを阻害する)J Antibiot (Tokyo). 2021 Oct;74(10):763-766.
【要旨の抜粋】
ニトロキソリンは、尿路感染症の治療に使用される金属キレート活性を持つ抗生物質である。 この小分子は、がんにおけるがん遺伝子の発現を調節するBET(bromodomain and extraterminal)タンパク質の機能も阻害する。Epstein-Barrウイルス (EBウイルス) によって引き起こされるリンパ球増殖は、BETタンパク質に依存する。 したがって、細胞培養およびEBウイルス関連リンパ球増殖症の小動物モデルに対するニトロキソリンの有効性をテストした。
ニトロキソリンは培養細胞と動物モデルの両方において腫瘍の増殖を抑える。 ニトロキソリンは、代表的な BET 阻害剤のJQ1 よりも速く効果を発揮する。 この合理的な医薬品転用は、臨床応用の可能性を秘めている。
【ニトロキソリンはBETタンパク質阻害を介してPD-L1の発現を抑制する】
岡山大学の泌尿器科のグループから以下のような論文が報告されています。
The Novel Combination of Nitroxoline and PD-1 Blockade, Exerts a Potent Antitumor Effect in a Mouse Model of Prostate Cancer.(ニトロキソリンとPD-1遮断薬の新規併用は前立腺がんのマウスモデルにおいて強力な抗腫瘍効果を発揮する)Int J Biol Sci. 2019 Mar 9;15(5):919-928.
【要旨】
プログラム細胞死タンパク質1(Programmed cell death protein 1:PD-1)遮断は前立腺がんに対する有望な治療戦略である。ニトロキソリンは、いくつかの種類のがんにおいて有効な抗がん作用を有することが知られている。前立腺がんのマウスの実験モデルにおけるニトロキソリンとPD 1遮断の併用療法の有効性を検討した。
インビトロの実験系において、ニトロキソリンはマウス前立腺がん細胞株RM9-Luc-PSAの生存と増殖を阻害することを見出した。
さらに、ニトロキソリンは、リン酸化PI3キナーゼ、リン酸化Akt(Thr308)、リン酸化Akt(Ser473)、リン酸化GSK-3β、Bcl-2、およびBcl-xLの発現を抑制した。
さらに、ニトロキソリンは培養した前立腺がん細胞および腫瘍組織におけるプログラム細胞死リガンド-1(PD-L1)の発現レベルを抑制した。
マウス前立腺がん同所性移植モデルにおいて、ニトロキソリン+ PD-1遮断は、ニトロキソリンまたはPD-1遮断をそれぞれ単独で使用した場合と比較して、腫瘍増殖を相乗的に抑制し、腫瘍重量、生物発光腫瘍シグナル、および血清中の前立腺特異抗原(PSA)レベルの減少をもたらした。
さらに、ニトロキソリンと PD-1遮断の併用は末梢血中のCD44+CD62L+CD8+ メモリーT細胞の細胞数の増加および骨髄由来抑制細胞の数を減少して、抗腫瘍免疫を有意に増強することを示した。
結論として、我々の実験結果はニトロキソリンとPD-1遮断薬の併用が、前立腺がん患者における有望な治療戦略になる可能性を示唆している。
ヒト型抗PD-1モノクローナル抗体のニボルマブ(nivolumab商品名「オプジーボ(Opdivo)」)はPD-1とPD-L1の結合を阻害することによって細胞傷害性T細胞の細胞死を防ぐ薬です。このような免疫チェックポイント阻害剤を使用すると、がん細胞を攻撃する細胞傷害性T細胞の働きを高めることが可能になります。免疫チェックポイント阻害剤にニトロキソリンを併用すると、抗腫瘍効果を高めることができるという報告です。
このPD-L1の発現抑制作用がBETタンパク質の阻害作用による可能性を示唆する研究が米国から報告されています。以下のような報告があります。
BET Bromodomain Inhibition Promotes Anti-Tumor Immunity by Suppressing PD-L1 expression.(BETブロモドメイン阻害はPD-L1発現を抑制することにより抗腫瘍免疫を促進する)Cell Rep. 2016 Sep 13; 16(11): 2829–2837.
【要旨】
この研究では、BETブロモドメイン阻害がPD-L1発現を抑制し、卵巣がんにおける腫瘍進行を抑制することを示す。
PD-L1をコードする遺伝子CD274は、BRD4媒介遺伝子転写の直接の標的である。
マウスの実験モデルにおいて、BET阻害剤のJQ1による治療は、腫瘍細胞ならびに腫瘍関連樹状細胞およびマクロファージにおけるPD-L1発現を有意に減少させ、これは抗腫瘍細胞傷害性T細胞の活性の増加と相関していた。 BET阻害剤は細胞傷害性T細胞依存的に腫瘍の進行を抑制した。
以上の結果は、PD-L1シグナル伝達を遮断するための化合物の有効性を実証している。 臨床試験でBET阻害剤は毒性が低く安全性が高いことが証明されているので、薬理学的BET阻害剤はPD-L1発現を標的とする治療戦略となり得ることを示している。
この論文では、BETファミリータンパク質のBRD4の阻害剤がPD-L1の発現を抑制することを報告しています。PD-L1をコードする遺伝子CD274は、BRD4媒介遺伝子転写の直接の標的だからです。
岡山大学泌尿器科のグループの論文では、ニトロキソリンがPD-L1の発現を阻害することを報告していますが、BRD4の関与は考察していません。しかし、前述のようにニトロキソリンはBETファミリータンパク質のBRD4を阻害する作用が報告されています。つまり、ニトロキソリンがBETブロモドメインの阻害によってPD-L1発現を抑制することが推測されます(下図)。

図:ヒストンのアセチル化されたリシンを認識するブロモドメインの繰り返し配列と特異的末端配列を持つBET(bromodomain and extra-terminal)ファミリータンパク質の一つのBRD4は、ヒストンのアセチル化リシンに結合し、転写因子やRNAポリメラーゼなどをリクルートして(①)、PD-L1をコードするCD274遺伝子の転写を促進する(②)。ニトロキソリンはBRD4の第一ブロモドメインとアセチル化ヒストンの結合を阻害する(③)。その結果、転写因子やRNAポリメラーゼのリクルートが阻害され(④)、CD274遺伝子の転写を抑制し、PD-L1の発現を抑制する(⑤)。
この研究ではマウスの実験ではニトロキソリンを1日に体重1kg当たり15mgを経口投与しています。この量は人間に換算すると2〜3mg/kg/日程度になります。一般にマウスの体重当たりのエネルギー消費量や薬物の代謝速度は人間の約7倍と言われています。したがって、15mg/kgの7分の1の用量が一つの目安となります(詳しくは293話参照)
ニトロキソリンの尿路感染症の治療に使う量は1日に500から750mgです。したがって、尿路感染症に使用する服用量で十分な抗腫瘍効果が期待できそうです。
ニトロキソリンが骨髄由来抑制細胞を減少することが報告されています。以下のような報告があります。
Nitroxoline inhibits bladder cancer progression by reversing EMT process and enhancing anti-tumor immunity.(ニトロキソリンは、上皮間葉転換プロセスを逆転させ、さらに抗腫瘍免疫を強化することにより、膀胱癌の進行を抑制する)J Cancer. 2020 Sep 23;11(22):6633-6641.
この報告では、C3H/He マウスの皮下に膀胱癌細胞を移植した実験モデルにおいて、ニトロキソリンは、腫瘍増殖の有意な阻害をもたらした。さらに、末梢血細胞中の骨髄由来抑制細胞の割合は、ニトロキソリンの治療後に大幅に減少した。
【駆虫薬のニクロサミドはSTAT-3活性を阻害してPD-L1の発現を阻害する】
免疫チェックポイント阻害剤の単独治療では抗腫瘍効果が弱いので、この効果を高めるための医薬品再利用の検討が行われています。駆虫薬のニクロサミドについて以下のような報告があります。
Niclosamide, an antihelmintic drug, enhances efficacy of PD-1/PD-L1 immune checkpoint blockade in non-small cell lung cancer.(駆虫薬であるニクロサミドは、非小細胞肺癌における PD-1/PD-L1 免疫チェックポイント遮断の有効性を高める)J Immunother Cancer. 2019 Sep 11;7(1):245.
【要旨の抜粋】
背景: PD-1/PD-L1 遮断薬は、一部の患者の予後を改善するが、多くの患者の免疫療法に対する反応は依然として満足のいくものでは無い。PD-1/PD-L1 遮断薬の抗腫瘍活性を高める方法を見つけ出すことが重要である。
方法: 非小細胞肺がん細胞株とマウスのモデルを使用して、ニクロサミドと PD-L1 遮断薬の組み合わせが腫瘍の増殖と T 細胞機能に及ぼす影響を検討した。非小細胞肺がん患者の腫瘍サンプルにおける PD-L1 と リン酸化STAT3(p-STAT3) 発現の関係、および患者の生存率との関係を調査した。
結果: 培養肺がん細胞を使ったin vitroの実験では、ニクロサミドはPD-L1 遮断の存在下で T 細胞によって媒介されるがん細胞溶解を増強した。ニクロサミドと PD-L1 抗体を投与したマウスは、腫瘍増殖が有意に遅延し、生存率の増加を示した。これは、腫瘍浸潤 T 細胞とグランザイム B 放出の増加と関連していた。
重要なことに、ニクロサミドが非小細胞肺がん細胞において濃度依存的および時間依存的にPD-L1の発現を減少させることを発見した。これは、PD-L1遺伝子のプロモーターへのp-STAT3結合の遮断と関連していた。
結論:非小細胞肺がんに対するPD-L1 抗体による抗腫瘍効果をニクロサミドが増強することは、in vitro および in vivo での実験において観察され、PD-L1 のプロモーターへの p-STAT3 結合の阻害、および最終的に PD-L1 発現の低下が関与していた。これらの結果は、ニクロサミドと PD-1/PD-L1 遮断薬の併用療法の有効性を示唆している。
この実験結果をまとめると、以下のような図になります。

図:JAK(Janus Kinase;ヤーヌスキナーゼ)はサイトカイン受容体のサブユニットとして存在し、チロシンをリン酸化するチロシンキナーゼ活性を持つ。IL-6や上皮成長因子(EGF)などの受容体が刺激されると(①)、JAKが活性化されてSTAT3がリン酸化される(②)。STAT3は不活性な状態では細胞質に存在し、JAK(ヤーヌスキナーゼ)などでチロシン705がリン酸化されると、STAT3二分子のSH2ドメインが、それぞれ他方の分子のリン酸化チロシンと相互作用することにより二量体を形成して核内に移行する(③)。核内に移行したSTAT3二量体は、標的となるDNAに結合する事で転写を活性化する(④)。STAT3はPD-L1遺伝子の発現を誘導する(⑤)。産生されたPD-L1は細胞膜に発現し(⑥)、活性化した細胞障害性T細胞(CTL)のPD-1と結合するとCTLの細胞死を誘導して免疫応答を阻害する(⑦)。ニクロサミドはSTAT3のリン酸化による活性化を阻害する(⑧)。
以上のような報告から、メラトニン、イベルメクチン、メトホルミン、ニトロキソリン、ニクロサミドの併用はPD-L1の発現を抑制することによって抗腫瘍免疫を相乗的に高める効果が期待できます。(トップの図)
これらはオプジーボなどの免疫チャックポイント阻害剤の効果を高めます。
さらに、ミトコンドリアの活性を高めるベザフィブラートがオプジーボの抗腫瘍効果を高めることが報告されています。
リンパ球の働きを弱めるがん組織の酸性化の改善(重曹などによるがん組織のアルカリ化)、骨髄由来抑制細胞の活性抑制などを併用すると、がん細胞を免疫システムで排除することが可能になります。
| « 833)抗腫瘍免... | 835)油を変え... » |