がんの予防や治療における漢方治療の存在意義を考察しています。がん治療に役立つ情報も紹介しています。
「漢方がん治療」を考える
カレンダー
| 2025年1月 | ||||||||
| 日 | 月 | 火 | 水 | 木 | 金 | 土 | ||
| 1 | 2 | 3 | 4 | |||||
| 5 | 6 | 7 | 8 | 9 | 10 | 11 | ||
| 12 | 13 | 14 | 15 | 16 | 17 | 18 | ||
| 19 | 20 | 21 | 22 | 23 | 24 | 25 | ||
| 26 | 27 | 28 | 29 | 30 | 31 | |||
|
||||||||
goo ブログ
過去の記事
カテゴリ
最新の投稿
最新のコメント
最新のトラックバック
ブックマーク
プロフィール
検索
gooおすすめリンク



| URLをメールで送信する | |
| (for PC & MOBILE) | |
800) がん細胞のPD-1(Programmed death-1)リガンド(PD-L1)の発現を抑制する方法(その2):メトホルミンとメラトニンとニトロキソリン
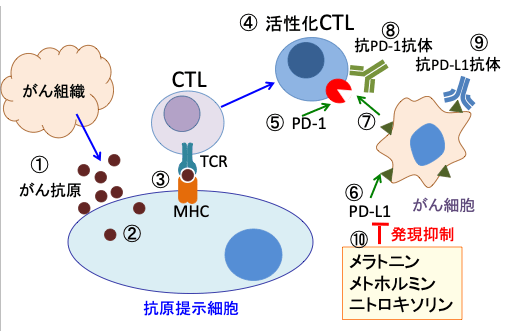
図:がん細胞から放出されたがん抗原(①)は、樹状細胞やマクロファージなどの抗原提示細胞に取込まれ(②)、ペプチドに分解されて抗原ペプチドとして抗原提示細胞上のMHC(major histocompatibility complex:主要組織適合抗原複合体)に提示される(③)。MHCはがん抗原を介してCTL(細胞傷害性T細胞)上のTCR(T細胞受容体)と反応してCTLを活性化する(④)。CTLは活性化されるとPD-1(Programmed death-1)が発現する(⑤)。がん細胞にはPD-1のリガンドであるPD-L1が発現している(⑥)。PD-1とPD-L1が結合するとCTLは増殖が抑制される(⑦)。PD-1とPD-L1の結合を抗PD-1抗体(⑧)や抗PD-L1抗体(⑨)で阻止するとCTLの抗腫瘍活性を高めることができる。メラトニン、メトホルミン、ニトロキソリンはがん細胞のPD-L1の発現量を減らす作用があり、CTLによる抗腫瘍効果を高めることができる(⑩)。
800) がん細胞のPD-1(Programmed death-1)リガンド(PD-L1)の発現を抑制する方法(その2):メトホルミンとメラトニンとニトロキソリン
【抗原提示とT細胞の活性化】
リンパ球のT細胞は、がん抗原で活性化されて初めて細胞傷害活性を持つようになります。すなわち、細胞傷害活性を持たないT細胞が抗原提示細胞(マクロファージや樹状細胞)から抗原ペプチド(がん抗原)を提示されて活性化してはじめてがん細胞に対して特異的な細胞傷害活性を持つ細胞傷害性T細胞(キラーT細胞)となり、がん細胞を攻撃するようになります。
細胞傷害性T細胞は細胞傷害物質であるパーフォリン、 グランザイム, TNF(tumor necrosis factor)などを放出したり、ターゲット細胞のFasを刺激してアポトーシスに陥らせることでがん細胞やウイルス感染細胞を死滅させます。
細胞傷害性T細胞の一部はメモリーT細胞となって、異物に対する細胞傷害活性を持ったまま宿主内に記憶され、次に同じ異物(抗原)に暴露された場合に対応できるよう備えます。
病原微生物が侵入したり、何らかの原因で炎症が起こると、血管から顆粒球や単球などが遊走して来ます。このように炎症反応によって集まってきたり、あるいは組織に常在していた樹状細胞やマクロファージは、侵入した細菌やウイルス粒子、あるいは死滅した細胞の死骸や断片などを取り込み、リンパ液の流れに沿って所属リンパ節に移動します。
樹状細胞やマクロファージは取り込んだタンパク質を分解し、その結果産生されたペプチド(アミノ酸が数個から数十個つながったもの)をMHC(major histocompatibility complex:主要組織適合抗原複合体)分子の上に提示します。
活性化した樹状細胞はリンパ節で手当たりしだいにナイーブT細胞(まだ一度も活性化されたことのないT細胞)とくっつきあって、何かを確かめます。ナイーブT細胞はその表面にT細胞抗原認識受容体(TCR)を持っています。樹状細胞の表面に提示されたMHC+抗原ペプチドとピタッとくっつく受容体(TCR)をもったナイーブT細胞と出会うと、そのT細胞を活性化します。
抗原を提示して活性化している樹状細胞にはCD80/86という補助刺激因子が発現しており、T細胞のCD28と結合し、刺激を送ります。
さらに、活性化した樹状細胞はサイトカインを放出しており、ナイーブT細胞はそれを浴びることになります。
このように、TCRを介するシグナルとCD28を介する補助刺激とサイトカインによる刺激を同時に受けたTリンパ球は初めて活性化し、TCRの特異性を保ったままで分裂・増殖して自らのクローンを増やします。
CD8陽性T細胞(キラーT細胞)は成熟し、細胞質内にパーフォリンやグランザイムなどを含んだ細胞傷害顆粒を持つエフェクター細胞になります。
エフェクター細胞はリンパ節を離れ、胸管を経て循環血液中へと流れ込み、血流に従って全身を巡ります。炎症の起こっている組織から産生されるサイトカインやケモカインなどの作用でエフェクターT細胞は炎症部位に集まり、病原菌やがん細胞の攻撃に参加します。

図:がん細胞から放出されたがん抗原を未熟樹状細胞が取り込んで成熟して抗原を提示するとき、MHC(major histocompatibility complex:主要組織適合抗原複合体)分子にペプチド抗原を載せて細胞傷害性T細胞やヘルパーT細胞に提示する。このとき、MCH+ペプチド抗原にぴったり結合するTCR(T細胞受容体)を持つT細胞は、補助刺激因子(CD28とCD80/86など)や樹状細胞から放出されるサイトカインの働きで活性化され、がん抗原を認識するT細胞がクローン性増殖(clonal expansion)し、がん細胞を攻撃する。
【抗がん剤や放射線は抗腫瘍免疫を刺激する】
放射線治療が全身の抗腫瘍免疫の活性化の引き金になりうることが明らかになっています。
がん組織に放射線照射を行うと、がん細胞が死滅して細胞内成分が放出されるとこれらの成分が危険シグナルとなって自然免疫が活性化されます。同時に死滅したがん細胞からがん抗原が放出され、このがん抗原の情報を抗原提示細胞(樹状細胞やマクロファージ)が細胞傷害性T細胞(CTL)に提示してCTLは活性化され、獲得免疫が成立すると、生き残ったがん細胞を攻撃して排除しようとします。このような非照射のがん細胞にも免疫細胞の作用が働くことをアブスコパル効果(Abscopal efffect)と言います。
同様に、抗がん剤治療も、分裂しているがん細胞を死滅させるだけでなく、この死滅した細胞から放出された細胞成分が自然免疫を刺激し、がん抗原が樹状細胞などの抗原提示細胞に認識されて、がん抗原特異的な抗腫瘍免疫を引き起こします(下図)。
したがって、通常の抗がん剤治療と免疫チェックポイント阻害剤の併用が相乗効果を示す可能性があり、実際に、抗がん剤と免疫チェックポイント阻害剤の併用による奏功率の向上が報告されています。

図:放射線治療や抗がん剤治療でがん細胞が死滅するとがん抗原が放出される(①)。がん抗原は樹状細胞やマクロファージなどの抗原提示細胞に取込まれ、ペプチドに分解されて抗原ペプチドとして抗原提示細胞上のMHC(主要組織適合抗原複合体)に提示される(②)。MHCはがん抗原を介してCTL(細胞傷害性T細胞)上のTCR(T細胞受容体)と反応してCTLを活性化し、抗原提示を受けたがん抗原特異的なCTLはクローン性に増殖し(③)、がん抗原を持っているがん細胞を攻撃する(④)。
【細胞傷害性T細胞を抑制するPD-1とCTLA-4】
リンパ球の一種のT細胞は、病原菌やがん細胞を攻撃・排除する働きがあります。しかし、T細胞が暴走して正常な細胞を攻撃すると危険なので、いくつかのブレーキ装置が備わっています。これを「免疫チェックポイント」と呼びます。
がん細胞は、ときに巧みにこの免疫チェックポイントを悪用して、T細胞にブレーキをかけてT細胞からの攻撃を逃れようとするのです。がん細胞によるブレーキがかからないようにする薬が免疫チェックポイント阻害薬です。
細胞傷害性T細胞(キラーT細胞)は抗原提示細胞(樹状細胞やマクロファージ)から抗原を提示されると活性化されて、敵(病原菌やがん細胞など)を攻撃します。
細胞傷害性T細胞にはPD-1やCTLA-4という受容体が存在します。PD-1はプログラム細胞死1(programmed death-1)、CTLA-4は細胞傷害性Tリンパ球抗原-4 (cytotoxic T-lymphocyte-associated protein 4)の略です。
これらの受容体のリガンド(受容体に結合して作用する物質)となるPD-L1やB7(B7-1, B7-2)を抗原提示細胞が持つことによって細胞傷害性T細胞の働きを抑制しています。
つまり、PD-1受容体やCTLA-4受容体がリガンドによって刺激されると、T細胞の増殖が停止し細胞死を来すことになります。このようにして細胞傷害性T細胞の過剰な応答を制御しています。
細胞傷害性T細胞の働きを阻害するPD-L1やB7はがん細胞にも発現しています。つまり、がん細胞は免疫系の制御システムを利用して、がん組織内の細胞傷害性T細胞の働きを阻止しています。
PD-1受容体やCTLA-4受容体は細胞傷害性T細胞を死滅させるスイッチなようなものなので、これらのスイッチが入らないようにすれば、細胞傷害性T細胞は生き残ってがん細胞の攻撃力を高めることができます。
CTLA-4に対する抗体(ヒト型抗ヒトCTLA-4モノクローナル抗体)のイピリブマブ(ipilimumab: YERVOY)やヒト型抗PD-1モノクローナル抗体のニボルマブ(nivolumab商品名「オプジーボ(Opdivo)」)などがあります。このような免疫チェックポイント阻害剤を使用すると、がん細胞を攻撃する細胞傷害性T細胞の働きを高めることが可能になります。
体に備わったがん細胞に対する攻撃力を高めてがんを治療しようというのが「がんの免疫療法」の理論です。「免疫細胞を活性化する」という従来の免疫療法では十分な効果が得られなかったのですが、その大きな理由は免疫応答にブレーキをかける仕組みの存在です。このブレーキを解除して免疫細胞に100%の力でがん細胞を攻撃させようというのが、CTLA-4やPD-1/PD-L1をターゲットにした治療法です。(下図)
ただ、この治療法は免疫細胞の暴走を許して、自己免疫疾患を引き起こすという副作用もあります。

図:抗原提示細胞上にはMHCクラスII(MHC-II)といわれる分子があり、抗原を介してT細胞上のTCR(T細胞受容体)と反応して細胞傷害性T細胞を活性化する(①)。T細胞上にはCD28とCTLA-4があり、CD28は恒常的に発現し、抗原提示細胞からのB7-1やB7-2というリガンドによってT細胞活性化に作用する(②)。一方、CTLA-4はT細胞活性化にともなって発現が誘導され、B7-1やB7-2によって刺激されるとT細胞を抑制する(③)。CTLA-4はCD28よりもB7に対する親和性が強いので、活性化したT細胞の過剰な応答を抑制する。同様に、PD-1(Programmed death-1)は抗原提示細胞のPD-L1(別名B7-H1)と結合することによって抑制型の免疫調節シグナルを活性化させる(④)。がん細胞もB7-1やB7-2やPD-L1が発現しており、細胞傷害性T細胞の働きを抑制している。T細胞のCTLA-4とPD-1の働きを特異抗体で阻害すると、がん細胞に対する細胞傷害性T細胞の働きを高めることができる(⑤)。
がん細胞を非自己と認識して、それを攻撃するためにT細胞は活性化しますが、PD-1リガンド(PD-L1)を持ったがん細胞と接触すると、CTL上のPD-1とリガンド(PD-L1)が結合することにより、免疫シグナルは抑制され、T細胞はがん細胞を攻撃できなくなってしまいます。これがT細胞を活性化するだけの従来の免疫療法に限界があった理由です。活性化したCTL(細胞傷害性T細胞)をがん組織に送っても、がん細胞を攻撃しようと近づくとPD-L1によって自身のPD-1のスイッチが入って死滅するからです。
がん細胞がPD-1リガンドを多く発現しているほど、予後が悪いというデータも報告されています。
T細胞やNK細胞を活性化すると同時に、T細胞上のPD-1やCTLA-4の働きをd阻止すると、がん組織を免疫力だけで縮小できる可能性があります。がん細胞に発現しているPD-1やCTLA-4のリガンドの発現量を減らす方法も有効です。
前回(799話)ではイベルメクチンとカンナビジオールがPAK1阻害を介して癌細胞のPD-L1の発現を抑制する効果があることを紹介しました。さらにメトホルミン、メラトニン、ニトロキソリンにもがん細胞のPD-L1の発現量を減らす効果が報告されています。
【メトホルミンは免疫チェックポイント阻害剤の効き目を高める】
以下のような報告があります。
Metformin Promotes Antitumor Immunity via Endoplasmic-Reticulum-Associated Degradation of PD-L1.(メトホルミンはPD-L1の小胞体関連の分解を介して抗腫瘍免疫を増強する)Mol Cell. 2018 Aug 16;71(4):606-620.
【要旨】
メトホルミンは抗腫瘍活性を有し、さらに細胞傷害性Tリンパ球による免疫監視機構を増強することが報告されている。ただし、がん免疫におけるメトホルミンの役割の機能と詳細なメカニズムは完全には理解されていない。ここでは、メトホルミンがプログラム細胞死リガンド-1(programmed death ligand-1:PD-L1)の安定性と膜局在を減らすことによって細胞傷害性Tリンパ球活性を高めることを示す。
さらに、メトホルミンによって活性化されたAMP活性化プロテインキナーゼ(AMPK)が直接PD-L1のセリン195をリン酸化することを発見した。 セリン195のリン酸化は異常なPD-L1のグリコシル化を誘導し、その結果、小胞体での蓄積および小胞体関連タンパク質分解を誘導した。
この結果と一致するように、メトホルミン治療を受けた乳がん患者の腫瘍組織は、AMPK活性化を伴うPD-L1レベルの低下を示した。メトホルミンによるPD-L1の阻害シグナルの遮断はがん細胞に対する細胞傷害性Tリンパ球活性を増強する。
この実験結果は小胞体関連タンパク質分解を介するPD-L1発現の新しい調節メカニズムを明らかにし、メトホルミンと免疫チェックポイント阻害剤の組み合わせが免疫療法の有効性を高める可能性があることを示唆している。
メトホルミンはオプジーボやキイトルーダなどの免疫チェックポイント阻害剤の抗腫瘍効果を高める効果が期待できると言うことです。以下のような報告もあります。
Efficacy of metformin in combination with immune checkpoint inhibitors (anti-PD-1/anti-CTLA-4) in metastatic malignant melanoma.(転移のある悪性黒色腫における免疫チェックポイント阻害剤(抗PD-1抗体/抗CTLA-4抗体)との併用におけるメトホルミンの効果)J Immunother Cancer. 2018 Jul 2;6(1):64. doi: 10.1186/s40425-018-0375-1.
【要旨】
背景:メトホルミンは、II型糖尿病の患者に一般的に使用されるビグアニドの1つである。 その血糖降下作用とは別に、メトホルミンはまた、LKB1 / AMPK経路に作用して、タンパク質合成および細胞増殖を抑制する。 さらに、腫瘍低酸素の減少によりPD-1阻害を増強する。 メトホルミンは、固形腫瘍における治療関連転帰に有意な好影響を示したが、これらの結果は、悪性黒色腫について行われた限られた臨床研究では再現されていない。 さらに、これらの研究のいずれも、メトホルミンと免疫チェックポイント阻害剤との併用の有効性に関する研究報告は無い。
方法:これは、2011年1月1日から2017年12月15日までに転移性悪性黒色腫と診断され、イピリムマブ(ipilimumab)、ニボルマブ(nivolumab)および/またはペンブロリズマブ(pembrolizumab)で治療された患者を含む後ろ向きコホート研究である。A群はこれらの免疫チョックポイント阻害剤のみで治療を受け、B群は免疫チェックポイント阻害剤にメトホルミンを併用した。主要エンドポイントは客観的反応率で、副次的エンドポイントは 疾患コントロール率、全生存期間および無増悪生存期間とした。
結果:A群は33人(60%)、B群は22人(40%)であった。 全患者の特徴は両コホート間で類似していた。 客観的反応率は、B群においてより高かった(68.2%対54.5%、P = 0.31)。 疾患コントロール率もB群の方が高かった(77.3%対60.6%、P = 0.19)。 B群では全生存期間中央値(46.7ヶ月対28ヶ月間)および無増悪生存期間中央値(19.8ヶ月対5ヶ月間)とも長かった。しかし、単変量および多変量解析では、これらの差異のいずれも統計的に有意ではなかった。 治療中に出現した新しい転移部位の平均数は、A群において有意に高かった(A:1.51対B:0.59、P = 0.009)。
結論:免疫チェックポイント阻害剤とメトホルミンの併用による治療は、免疫チェックポイント阻害剤単独の治療より良好な治療関連アウトカム(客観的反応率、疾患コントロール率、無増悪生存期間および全生存期間)が観察された。統計的有意差に達しなかったが、これはサンプルサイズが小さかったためと思われる。 したがって、免疫チャックポイント阻害剤とメトホルミンの併用が標準的な併用療法として推奨される前に、大きな前向き臨床試験が必要である。
【メラトニンはPD-L1の発現を抑制する】
以下のような報告があります。
Melatonin Downregulates PD-L1 Expression and Modulates Tumor Immunity in KRAS-Mutant Non-Small Cell Lung Cancer.(メラトニンはKRAS変異型非小細胞肺がんのPD-L1発現を低下し、腫瘍免疫を調節する)Int. J. Mol. Sci. 2021, 22(11), 5649
【要旨】
KRAS変異を有する非小細胞肺がん患者は、化学療法や免疫療法に対して抵抗性を示す。KRAS変異非小細胞肺がんに対する免疫療法の効果を高める戦略が求められている。
メラトニンは幅広い抗がん作用を示しているが、抗腫瘍免疫の調節にメラトニンの効果はほとんど知られていない。本研究では、メラトニンは、A549、H460、およびLLC1細胞を含むKRAS変異の非小細胞性肺がん細胞株で細胞アポトーシスを誘導し、細胞生存率を大幅に低下させた。
KRAS変異を有する肺がん細胞は、PD-L1(programmed death ligand 1)の発現の亢進が認められた。メラトニンによる治療は、インターフェロン(IFN)-γ刺激の存在下と非存在下の両方でPD-L1発現を低下した。
さらに、KRAS変異肺がん細胞はより高いYes関連タンパク質(YAP)およびPDZ結合モチーフ(TAZ)レベルの転写共活性化因子を示し、PD-L1発現は肺がん細胞のYAPおよびTAZと正の相関が認められた。
メラトニンによる治療は、YAP/TAZ下流遺伝子発現のダウンレギュレーションを伴うYAPおよびTAZを効果的に抑制した。メラトニンとYAP/TAZ阻害剤の組み合わせは、YAPとPD-L1の発現を確実に減少させた。
公開されたデータベースを使用した臨床分析により、PD-L1の発現は肺がん患者のYAPおよびTAZと正の相関があり、PD-L1の過剰発現は生存確率が低いことが示唆された。動物実験はさらに、メラトニンの投与が同系マウスモデルにおいて腫瘍増殖を有意に阻害し、腫瘍免疫を調節することを明らかにした。
以上から、これらのデータは、YAP / PD-L1軸を抑制することによって免疫抑制腫瘍微小環境を調節するメラトニンの新しい抗腫瘍メカニズムを明らかにし、非小細胞性肺がんを治療するためのメラトニンの治療可能性を示唆している。
YAP(Yes-associated protein)とTAZ(transcriptional co-activator with PDZ-binding motif) は転写共役因子です。
YAP/TAZは、細胞増殖や組織傷害後の組織再生を促進する遺伝子の発現を亢進します。したがって、再生医学ではYAP/TAZの活性化が治療に使われます。
しかし、がん細胞においては、アポトーシス抵抗性、増殖促進、幹細胞の性状(stemness)に関与する遺伝子の発現を亢進するので、YAP/TAZはがん細胞の増殖を促進することになり、YAP/TAZの活性化を阻害することが治療になります。
多くのがん細胞でYAP/TAZの活性が亢進しています。変異KRASがYAPを活性化して発がんを引き起こすことが報告されています。この論文ではKRASの変異のある非小細胞性肺がんでYAP/TAZの活性化が起こっていることを明らかにしています。
メラトニンはYAP/TAZの活性を抑制してPD-L1の発現を低下させる効果があるようです。イベルメクチンもYAP/TAZの活性を抑制します。
【ニトロキソリンはBETタンパク質阻害を介してPD-L1の発現を抑制する】
岡山大学の泌尿器科のグループから以下のような論文が報告されています。
The Novel Combination of Nitroxoline and PD-1 Blockade, Exerts a Potent Antitumor Effect in a Mouse Model of Prostate Cancer.(ニトロキソリンとPD-1遮断薬の新規併用は前立腺がんのマウスモデルにおいて強力な抗腫瘍効果を発揮する)Int J Biol Sci. 2019 Mar 9;15(5):919-928.
【要旨】
プログラム細胞死タンパク質1(Programmed cell death protein 1:PD-1)遮断は前立腺がんに対する有望な治療戦略である。ニトロキソリンは、いくつかの種類のがんにおいて有効な抗がん作用を有することが知られている。前立腺がんのマウスの実験モデルにおけるニトロキソリンとPD 1遮断の併用療法の有効性を検討した。
インビトロの実験系において、ニトロキソリンはマウス前立腺がん細胞株RM9-Luc-PSAの生存と増殖を阻害することを見出した。
さらに、ニトロキソリンは、リン酸化PI3キナーゼ、リン酸化Akt(Thr308)、リン酸化Akt(Ser473)、リン酸化GSK-3β、Bcl-2、およびBcl-xLの発現を抑制した。
さらに、ニトロキソリンは培養した前立腺がん細胞および腫瘍組織におけるプログラム細胞死リガンド-1(PD-L1)の発現レベルを抑制した。
マウス前立腺がん同所性移植モデルにおいて、ニトロキソリン+ PD-1遮断は、ニトロキソリンまたはPD-1遮断をそれぞれ単独で使用した場合と比較して、腫瘍増殖を相乗的に抑制し、腫瘍重量、生物発光腫瘍シグナル、および血清中の前立腺特異抗原(PSA)レベルの減少をもたらした。
さらに、ニトロキソリンと PD-1遮断の併用は末梢血中のCD44+CD62L+CD8+ メモリーT細胞の細胞数の増加および骨髄由来抑制細胞の数を減少して、抗腫瘍免疫を有意に増強することを示した。
結論として、我々の実験結果はニトロキソリンとPD-1遮断薬の併用が、前立腺がん患者における有望な治療戦略になる可能性を示唆している。
PD-1受容体は細胞傷害性T細胞を死滅させるスイッチなようなものなので、このスイッチが入らないようにすれば、細胞傷害性T細胞は生き残ってがん細胞の攻撃力を高めることができます。
ヒト型抗PD-1モノクローナル抗体のニボルマブ(nivolumab商品名「オプジーボ(Opdivo)」)はPD-1とPD-L1の結合を阻害することによって細胞傷害性T細胞の細胞死を防ぐ薬です。このような免疫チェックポイント阻害剤を使用すると、がん細胞を攻撃する細胞傷害性T細胞の働きを高めることが可能になります。免疫チェックポイント阻害剤にニトロキソリンを併用すると、抗腫瘍効果を高めることができるという報告です。
このPD-L1の発現抑制作用がBETタンパク質の阻害作用による可能性を示唆する研究が米国から報告されています。以下のような報告があります。
BET Bromodomain Inhibition Promotes Anti-Tumor Immunity by Suppressing PD-L1 expression.(BETブロモドメイン阻害はPD-L1発現を抑制することにより抗腫瘍免疫を促進する)Cell Rep. 2016 Sep 13; 16(11): 2829–2837.
【要旨】
抗体を用いてPD-L1シグナル伝達を遮断することによる抗腫瘍免疫の回復はがん治療に有益であることが証明されている。
この研究では、BETブロモドメイン阻害がPD-L1発現を抑制し、卵巣がんにおける腫瘍進行を抑制することを示す。
PD-L1をコードする遺伝子CD274は、BRD4媒介遺伝子転写の直接の標的である。
マウスの実験モデルにおいて、BET阻害剤のJQ1による治療は、腫瘍細胞ならびに腫瘍関連樹状細胞およびマクロファージにおけるPD-L1発現を有意に減少させ、これは抗腫瘍細胞傷害性T細胞の活性の増加と相関していた。 BET阻害剤は細胞傷害性T細胞依存的に腫瘍の進行を抑制した。
以上の結果は、PD-L1シグナル伝達を遮断するための化合物の有効性を実証している。 臨床試験でBET阻害剤は毒性が低く安全性が高いことが証明されているので、薬理学的BET阻害剤はPD-L1発現を標的とする治療戦略となり得ることを示している。
この論文では、BETファミリータンパク質のBRD4の阻害剤がPD-L1の発現を抑制することを報告しています。PD-L1をコードする遺伝子CD274は、BRD4媒介遺伝子転写の直接の標的だからです。
岡山大学泌尿器科のグループの論文では、ニトロキソリンがPD-L1の発現を阻害することを報告していますが、BRD4の関与は考察していません。しかし、ニトロキソリンはBETファミリータンパク質のBRD4を阻害する作用が報告されています。(657話参照)
つまり、ニトロキソリンがBETブロモドメインの阻害によってPD-L1発現を抑制することが推測されます(下図)。

図:ヒストンのアセチル化されたリシンを認識するブロモドメインの繰り返し配列と特異的末端配列を持つBET(bromodomain and extra-terminal)ファミリータンパク質の一つのBRD4は、ヒストンのアセチル化リシンに結合し、転写因子やRNAポリメラーゼなどをリクルートして(①)、PD-L1をコードするCD274遺伝子の転写を促進する(②)。ニトロキソリンはBRD4の第一ブロモドメインとアセチル化ヒストンの結合を阻害する(③)。その結果、転写因子やRNAポリメラーゼのリクルートが阻害され(④)、CD274遺伝子の転写を抑制し、PD-L1の発現を抑制する(⑤)。
この研究ではマウスの実験ではニトロキソリンを1日に体重1kg当たり15mgを経口投与しています。この量は人間に換算すると2〜3mg/kg/日程度になります。一般にマウスの体重当たりのエネルギー消費量や薬物の代謝速度は人間の約7倍と言われています。したがって、15mg/kgの7分の1の用量が一つの目安となります(詳しくは293話参照)
ニトロキソリンの尿路感染症の治療に使う量は1日に500から750mgです。したがって、尿路感染症に使用する服用量で十分な抗腫瘍効果が期待できそうです。
以上のような報告から、イベルメクチン、メトホルミン、メラトニンなどによるPD-L1発現抑制にニトロキソリンの併用はがん治療に相乗効果が期待できます。
| « 799) がん細胞... | 801)ドコサヘ... » |