がんの予防や治療における漢方治療の存在意義を考察しています。がん治療に役立つ情報も紹介しています。
「漢方がん治療」を考える
カレンダー
| 2025年1月 | ||||||||
| 日 | 月 | 火 | 水 | 木 | 金 | 土 | ||
| 1 | 2 | 3 | 4 | |||||
| 5 | 6 | 7 | 8 | 9 | 10 | 11 | ||
| 12 | 13 | 14 | 15 | 16 | 17 | 18 | ||
| 19 | 20 | 21 | 22 | 23 | 24 | 25 | ||
| 26 | 27 | 28 | 29 | 30 | 31 | |||
|
||||||||
goo ブログ
過去の記事
カテゴリ
最新の投稿
最新のコメント
最新のトラックバック
ブックマーク
プロフィール
検索
gooおすすめリンク



| URLをメールで送信する | |
| (for PC & MOBILE) | |
799) がん細胞のPD-1(Programmed death-1)リガンド(PD-L1)の発現を抑制する方法(その1):イベルメクチンとカンナビジオールのPAK-1阻害作用
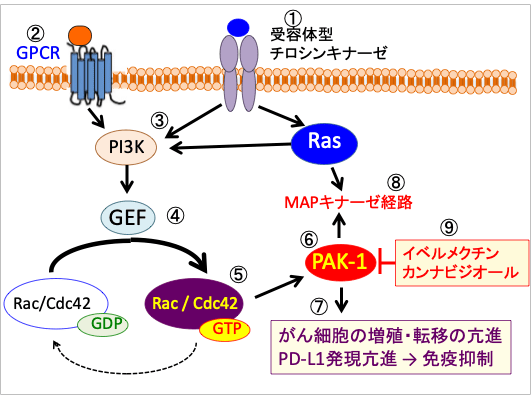
図:受容体型チロシンキナーゼ(インスリン受容体、IGF-1受容体、EGF受容体、PDGF受容体、VEGF受容体など)およびGタンパク質共役型受容体(G protein coupled receptor : GPCR)からのシグナル(①と②)は、RASおよびPI3Kを活性化し(③)、GDP/GTP交換反応を促進するGEF(guanine-nucleotide exchange factors)の作用によって(④)、Rac/Cdc42はGTP結合型になって活性化し(⑤)、Rac/Cdc42のエフェクターであるp21活性化キナーゼ(PAK-1)を活性化する(⑥)。活性化したPAK-1はがん細胞の増殖と転移を亢進し、PD-1(Programmed death-1)リガンド(PD-L1)の発現を亢進する(⑦)。さらに、PAK-1はRASによるMAPキナーゼ(分裂促進因子活性化タンパク質キナーゼ)経路の活性化を増強する(⑧)。イベルメクチンとカンナビジオールはPAK-1活性を阻害する作用がある(⑨)。
799) がん細胞のPD-1(Programmed death-1)リガンド(PD-L1)の発現を抑制する方法(その1):イベルメクチンとカンナビジオールのPAK-1阻害作用
【細胞傷害性T細胞を抑制するPD-1とCTLA-4】
細胞傷害性T細胞(キラーT細胞)は抗原提示細胞(樹状細胞やマクロファージ)から抗原を提示されると活性化されて、敵(病原菌やがん細胞など)を攻撃します。
細胞傷害性T細胞にはPD-1やCTLA-4という受容体が存在します。PD-1はプログラム細胞死1(programmed death-1)、CTLA-4は細胞傷害性Tリンパ球抗原-4 (cytotoxic T-lymphocyte-associated protein 4)の略です。
これらの受容体のリガンド(受容体に結合して作用する物質)となるPD-L1やB7(B7-1, B7-2)を抗原提示細胞が持つことによって細胞傷害性T細胞の働きを抑制しています。
つまり、PD-1受容体やCTLA-4受容体がリガンドによって刺激されると、T細胞の増殖が停止し細胞死を来すことになります。このようにして細胞傷害性T細胞の過剰な応答を制御しています。
細胞傷害性T細胞の働きを阻害するPD-L1やB7はがん細胞にも発現しています。つまり、がん細胞は免疫系の制御システムを利用して、がん組織内の細胞傷害性T細胞の働きを阻止しています。
PD-1受容体やCTLA-4受容体は細胞傷害性T細胞を死滅させるスイッチなようなものなので、これらのスイッチが入らないようにすれば、細胞傷害性T細胞は生き残ってがん細胞の攻撃力を高めることができます。
CTLA-4に対する抗体(ヒト型抗ヒトCTLA-4モノクローナル抗体)のイピリブマブ(ipilimumab: YERVOY)やヒト型抗PD-1モノクローナル抗体のニボルマブ(nivolumab商品名オプジーボ(Opdivo))などがあります。
このような免疫チェックポイント阻害剤を使用すると、がん細胞を攻撃する細胞傷害性T細胞の働きを高めることが可能になります。
体に備わったがん細胞に対する攻撃力を高めてがんを治療しようというのが「がんの免疫療法」の理論です。「免疫細胞を活性化する」という従来の免疫療法では十分な効果が得られなかったのですが、その大きな理由は免疫応答にブレーキをかける仕組みの存在です。このブレーキを解除して免疫細胞に100%の力でがん細胞を攻撃させようというのが、CTLA-4やPD-1/PD-L1をターゲットにした治療法です。(下図)
ただ、この治療法は免疫細胞の暴走を許して、自己免疫疾患を引き起こすという副作用もあります。

図:抗原提示細胞上にはMHCクラスII(MHC-II)といわれる分子があり、抗原を介してT細胞上のTCR(T細胞受容体)と反応して細胞傷害性T細胞を活性化する(①)。T細胞上にはCD28とCTLA-4があり、CD28は恒常的に発現し、抗原提示細胞からのB7-1やB7-2というリガンドによってT細胞活性化に作用する(②)。一方、CTLA-4はT細胞活性化にともなって発現が誘導され、B7-1やB7-2によって刺激されるとT細胞を抑制する(③)。CTLA-4はCD28よりもB7に対する親和性が強いので、活性化したT細胞の過剰な応答を抑制する。同様に、PD-1(Programmed death-1)は抗原提示細胞のPD-L1(別名B7-H1)と結合することによって抑制型の免疫調節シグナルを活性化させる(④)。がん細胞もB7-1やB7-2やPD-L1が発現しており、細胞傷害性T細胞の働きを抑制している。T細胞のCTLA-4とPD-1の働きを特異抗体で阻害すると、がん細胞に対する細胞傷害性T細胞の働きを高めることができる(⑤)。
がん細胞を非自己と認識して、それを攻撃するためにT細胞は活性化しますが、PD-1リガンド(PD-L1)を持ったがん細胞と接触すると、CTL上のPD-1とリガンド(PD-L1)が結合することにより、免疫シグナルは抑制され、T細胞はがん細胞を攻撃できなくなってしまいます。これがT細胞を活性化するだけの従来の免疫療法に限界があった理由です。活性化したCTL(細胞傷害性T細胞)をがん組織に送っても、がん細胞を攻撃しようと近づくとPD-L1によって自身のPD-1のスイッチが入って死滅するからです。
がん細胞がPD-1リガンドを多く発現しているほど、予後が悪いというデータも報告されています。
T細胞やNK細胞を活性化すると同時に、T細胞上のPD-1やCTLA-4の働きを阻止すると、がん組織を免疫力だけで縮小できる可能性が高まります。
がん細胞で活性化しているPAK-1(p21活性化キナーゼ-1)がPD-L1の発現を亢進するので、PAK-1の阻害がPD-L1の発現を抑制して、免疫チェックポイント阻害剤などのがんの免疫療法の効き目を高めることが報告されています。
【細胞は脂質二重層で包まれている】
体を構成する個々の細胞は細胞膜で囲まれています。細胞膜は脂質二重層によりできており、この細胞膜によって細胞外と細胞内が分けられています。脂質二重層はリン脂質分子が膜状に並んで作られます。リン脂質分子は親水性のリン酸部分と、疎水性の2個の脂肪酸が尻尾のように繋がった構造をしています。
細胞の内外は主に水で満たされているので、リン脂質分子は親水性のリン酸部分(頭部)を外側に、水に反発する疎水性の脂肪酸部分(尾部)を内側にして、7.5ナノメートル(nm)程度の厚さの2重の層を作って並びます(図)。

図:リン脂質は親水性のリン酸部分(頭部)と、疎水性の脂肪酸部分(尾部)から構成される。疎水性の尾部は水によってはじかれ、互いに引き付けられて内側に並び、親水性の頭部の領域が水に接する外側に露出して膜状の二重層を形成する。この脂質二重層が細胞膜の基本構造になる。
細胞の内外を分ける細胞膜は脂質二重層を土台にして、その中にタンパク質粒子が浮遊するように移動しています。脂質二重層に浮かぶタンパク質粒子は、細胞外からのシグナルを受け取る受容体や、物質を通すチャネルなどとして働きます。細胞膜に埋め込まれたタンパク質や脂質に糖鎖が結合し、細胞の識別や情報交換のマーカーとして細胞機能に影響を与えています。(図)
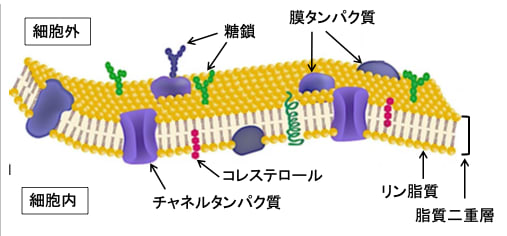
図:細胞膜は脂質の二重膜の海に、膜タンパク質が氷山のように頭を少し出して浮かんだような構造をしている。この構造モデルを流動モザイクモデルと呼んでいる。
【ホスファチジルイノシトール・シグナル伝達系】
ホスファチジルイノシトール(Phosphatidylinositol) は、イノシトール環と、グリセロール骨格を介して結合した2つの脂肪酸鎖からなる小さな脂質分子であり、細胞膜の細胞質面にアンカーされた構造を有します。

図:細胞膜はリン脂質の二重層で構成される。ホスファチジルイノシトールは負に帯電したリン脂質であり、リン脂質の微量成分を構成する。ホスファチジルイノシトールは、脂質二重層の細胞質側の脂質層に存在する。ホスファチジルイノシトールは、膜脂質二重層に埋め込まれた脂肪アシル鎖で構成され、グリセロール骨格を介して細胞質に面するミオイノシトール環に結合している。
ホスファチジルイノシトールは、多数の脂質キナーゼによってイノシトール環の3、4および/または5ヒドロキシ基がリン酸化されます。これにより、多様性に富むPhosphatidylinositol monophosphate (PI3P、PI4P、PI5P)、diphosphate [PI(3,4)P2、PI(3,5)P2 、PI(4,5)P2]、triphosphate [PI(3,4,5)P3]が産生され、これらを一まとめにしてホスフォイノシチド(Phosphoinositide)と呼ばれます。

図:ホスファチジルイノシトール(Phosphatidylinositol) は、イノシトール環と、グリセロール骨格を介して結合した2つの脂肪酸鎖から構成される。ホスファチジルイノシトールは、多数の脂質キナーゼによってイノシトール環の3、4および/または5ヒドロキシ基がリン酸化され、多様性に富むホスフォイノシチド(Phosphoinositide)が生成される。
生存、増殖、分化、DNA損傷応答および遺伝子転写を含む複数の細胞機能は、脂質セカンドメッセンジャーのホスファチジルイノシトールを調節するキナーゼ、ホスファターゼ、およびリパーゼのネットワークによって制御されています。
ホスファチジルイノシトールは、ホスホジエステル結合を介してグリセロールに結合した親水性のイノシトール部分と、疎水性の脂肪酸から構成されます。脂肪酸の部分は主にステアリン酸とアラキドン酸が結合しています。
ホスホリパーゼCによるホスホジエステル結合の加水分解とともに、リン酸基の付加または除去によるイノシトール環の修飾は、多くの細胞においてセカンドメッセンジャーとして働きを担っています。
【IP3はセカンドメッセンジャーとして細胞機能の制御に関わる】
真核細胞は、myo-イノシトール骨格を利用して、多様なシグナル伝達分子を生成しています。これは、6炭素のイノシトール環の周りにリン酸基を配置することによって生成されています。
イノシトールリン酸は、20世紀初頭に、主要なリン酸貯蔵庫を構成する植物の豊富な成分としてイノシトールヘキサキスリン酸(IP6、フィチン酸としても知られる)が同定されて以来、生物学的に重要な分子として認識されてきました。
これらの物質への関心は、1980年代半ばにIP3(イノシトール1,4,5-三リン酸)が細胞内貯蔵からカルシウムを放出するセカンドメッセンジャーとして発見されたことで高まりました。
これまでに最もよく研究されてきたPhosphoinositideはPI(3,4,5)P3であり、PI3K(ホスファチジルイノシトール3-キナーゼ)によってPI(4,5)P2から合成され、PTENにより脱リン酸化されます。
ホスファチジルイノシトール3-キナーゼ(PI3K)は、細胞膜の構成要素でホスホイノシチドの代謝前駆体であるホスファチジルイノシトールの3'-水酸基をリン酸化するシグナル伝達タンパク質で、広範な細胞活性を調節することによって細胞生存および細胞死を制御する生物活性脂質分子です。
PI3KおよびPTENは両方とも、受容体チロシンキナーゼに誘導されるAktシグナル伝達の中心的なメディエーターであり、これらの変異は多くのがん細部で高頻度に認められます。
ホスファチジルイノシトール3-キナーゼ(PI3K)は,細胞膜の構成成分であるイノシトールリン脂質をリン酸化する酵素です。ホスファチジルイノシトール4,5-二リン酸(PIP2)の3位のOHをリン酸化してホスファチジルイノシトール3,4,5-三リン酸(PIP3)を生成する酵素です。PIP3がAktをリン酸化して活性化します。
PTEN(Phosphatase and Tensin Homolog Deleted from Chromosome 10)はイノシトールリン脂質であるホスファチジルイノシトール-3,4,5-三リン酸(PIP3)の脱リン酸化反応を触媒する酵素です。PIP3をPIP2に変化することによってAktの活性化を阻止します。

図:インスリン、インスリン様成長因子-1(IGF-1)、成長ホルモンなどの増殖刺激が細胞に作用すると(①)、それらの受容体を介してPI3キナーゼ(PI3K)というリン酸化酵素が活性化される(②)。PI3Kは細胞膜の構成成分であるイノシトールリン脂質をリン酸化する酵素で,ホスファチジルイノシトール4,5-二リン酸(PIP2)の3位のOHをリン酸化してホスファチジルイノシトール3,4,5-三リン酸(PIP3)を生成する(③)。生成したPI3,4,5-三リン酸(PIP3)がAktをリン酸化して活性化する(④)。PTENは脱リン酸化する酵素でPIP3をPIP2に変化することによってAktの活性化を阻止する(⑤)。活性化したAktは、細胞内のシグナル伝達に関与する様々なタンパク質の活性を調節することによって細胞の増殖や生存(あるいは死)の調節を行う。がん細胞においてはAktの活性化はがん細胞の増殖・転移を亢進し、アポトーシスに抵抗性にする(⑥)。
PI3キナーゼ/AKT経路は,細胞外からのシグナルを細胞内に伝える主要な経路の一つで、増殖因子やインテグリンを介した細胞接着など、様々な刺激により活性化され、細胞の生存促進,細胞増殖・大きさの制御、細胞運動、代謝の制御など多くの細胞機能に関与しています。
ホスファチジルイノシトール3-キナーゼ(PI3K)は,細胞膜のホスファチジルイノシトール4,5-二リン酸(PIP2)の3位のOHをリン酸化してホスファチジルイノシトール3,4,5-三リン酸(PIP3)を生成し、生成したPI3,4,5-三リン酸(PIP3)がAktをリン酸化します。
Aktは多くのがん組織で活性化しておりAKTシグナル伝達系は、細胞増殖や生存、細胞サイズや栄養素利用への応答性、グルコース代謝、組織浸潤および血管新生など多くの細胞プロセスを制御しています。
がん細胞の生存と増殖はAKTシグナル伝達系の活性に依存度が高いので、AKTシグナル伝達系の阻害はがん細胞の増殖抑制やアポトーシス誘導を引き起こします。
PTENは脱リン酸化する酵素でPIP3をPIP2に変化することによってAktの活性化を阻止します。多くのがん細胞でPTENの変異が起こっています。
【がん細胞ではRAS変異によってp21活性化キナーゼ(PAK)が活性化している】
正常細胞は、増殖や細胞死や移動は厳密に制御されており、がん細胞はその制御が壊れて、無制限に増殖し、正常組織に浸潤し破壊して、増大していきます。細胞骨格はこれらのプロセスに必須の役割を担っています。
細胞の運動性を制御する細胞骨格の動的変化の背後にあるシグナル伝達経路にはRas関連の低分子量GTAアーゼ(Ras-related small GTPases)と、p21活性化キナーゼ(p21-activated kinases :PAKs)を含むエフェクタータンパク質が関与しています。
PAKキナーゼは酵母やショウジョウバエにも存在するセリン/スレオニンプロテインキナーゼのファミリーであり、哺乳類では6つのアイソフォーム(PAK1〜6)が発見されています。それらはすべて低分子量GTPaseの RacおよびCdc42の直接の標的です。
細胞骨格の動的変化の制御における役割に加えて、PAKは細胞の生存、分裂、遺伝子転写などの様々な細胞活動を調節することが明らかになっています。
いくつかの成長因子受容体チロシンキナーゼ(インスリン、IGF-1、EGF、PDGF、VEGF受容体など)およびGタンパク質共役受容体からのシグナルは、PAKの活性化につながります。
これらの経路は、PI-3キナーゼ(PI3K)とグアニンヌクレオチド交換因子(GEF)の連続的な活性化を通じて低分子量GTPase のRacおよびCdc42を活性化し、PAKを活性化します。
がん細胞では、PAKの活性化は変異Rasを介して頻繁に起こっています。
Rasは最も一般的に変異しているがん遺伝子の1つであり、MAPキナーゼ経路とPI3キナーゼを活性化し、PAKキナーゼを活性化します。

図:受容体チロシンキナーゼ(インスリン受容体、IGF-1受容体、EGF受容体、PDGF受容体、VEGF受容体など)およびGタンパク質共役型受容体(G protein coupled receptor : GPCR)からのシグナル(①と②)は、RASおよびPI3Kを活性化し(③)、GDP/GTP交換反応を促進するGEF(guanine-nucleotide exchange factors)の作用によって(④)、Rac/Cdc42はGTP結合型になって活性化し(⑤)、Rac/Cdc42のエフェクターであるp21活性化キナーゼ(PAK-1)を活性化する(⑥)。活性化したPAK-1はがん細胞の増殖と転移を亢進し、PD-L1の発現を亢進して抗腫瘍免疫を抑制する(⑦)。さらに、PAK-1はRASによるMAPキナーゼ(分裂促進因子活性化タンパク質キナーゼ)経路の活性化を増強する(⑧)。
【PAK-1はPD-L1の発現を亢進する】
以下のような報告があります。
Inhibition of PAK1 suppresses pancreatic cancer by stimulation of anti-tumour immunity through down-regulation of PD-L1. (PAK1の阻害は、PD-L1のダウンレギュレーションによる抗腫瘍免疫の刺激により、膵臓がんを抑制する)Cancer Lett. 2020 Mar 1;472:8-18.
【要旨】
膵臓がんに対する免疫療法は、活性化した膵星細胞による結合組織の増生などによる免疫抑制性の微小環境の存在のため、優位な臨床的利益をもたらしていない。
この研究は、抗腫瘍免疫におけるPAK1の関与を調査することを目的としている。
膵臓がん患者では、PAK1の発現が低く、膵星細胞の活性化が低く、CD8 + T細胞/PAK1の比率が高いことは、全生存期間の延長と相関していた。
マウスの膵臓がんのモデルでは、PAK1遺伝子のノックアウトにより腫瘍組織内のCD4+ T細胞およびCD8+ T細胞が増加し、膵臓星細胞の活性化が阻害され、生存期間が延長した。
PAK1の阻害は、膵星細胞の刺激による膵臓がん細胞の増殖と遊走を阻害し、細胞傷害性リンパ球による膵臓がん細胞の細胞死誘導に対する膵星細胞による保護作用を阻止し、膵臓がん細胞における内因性および星細胞刺激によるPD-L1発現を減少させ、膵臓がん細胞の細胞傷害性リンパ球による細胞死を増加した。
PAK1の阻害は、腫瘍組織内のCD4 +およびCD8 + T細胞を増加させ、内因性および星細胞誘導性のPD-L1発現のダウンレギュレーションを介して細胞傷害性リンパ球による殺傷に対してPDA細胞を感作することにより、抗腫瘍免疫を刺激します。
特に免疫チェックポイント阻害剤と組み合わせたPAK1阻害剤は、膵臓がんの免疫療法の有効性を改善する可能性があります。
【イベルメクチンはPAK-1を阻害する】
イベルメクチン(ivermectin)はマクロライド類に属する物質で、腸管糞線虫症や糸状虫や疥癬など多くの寄生虫に有効です。
静岡県伊東市内のゴルフ場近くで採取した土壌から大村智博士により発見された新種の放線菌「ストレプトマイセス・アベルメクチニウス」(Streptomyces avermitilis)が生産するアベルメクチンを元に創製されました。大村智博士はこの発見で2015年にノーベル生理学・医学賞を受賞しています。
イベルメクチンがPAK-1の活性を阻害することが報告されています。
以下のような報告があります。PAK研究の第一人者の丸田浩博士たちの研究報告です。
Ivermectin inactivates the kinase PAK1 and blocks the PAK1-dependent growth of human ovarian cancer and NF2 tumor cell lines.(イベルメクチンは、キナーゼPAK1を不活性化し、ヒト卵巣がんおよびNF2腫瘍細胞株のPAK1依存性の成長を阻止する)Drug Discov Ther. 2009 Dec;3(6):243-6.
【要旨】
イベルメクチンは、線虫のGABA(γ-アミノ酪酸)受容体を阻害し、哺乳類の対応する受容体には作用しないので、線虫を非常に低用量(0.2 mg / kg)で選択的に死滅する寄生虫治療薬である。
数年前にロシアのグループから、イベルメクチンは、マウスに悪影響を与えることなく、はるかに高い用量(3-5 mg / kg)でマウスのヒトメラノーマおよび他のいくつかのがん異種移植片の成長をほぼ完全に抑制できることが報告された。しかし、その抗がん機序は分子レベルでまだ解明されていない。
イベルメクチンのPAK1阻害作用の可能性に対する最初のヒントは、致死量以下のイベルメクチンが線虫の産卵数を劇的に減少させるという最近の発見であった。
PAK1遺伝子欠損(遺伝子ノックアウト)、またはプロポリスの主要な抗がん成分であるCAPE(カフェ酸フェネチルエステル)やARC(アルテピリンC)などの天然のPAK1阻害剤による治療も、全く同じ効果を引き起こす。この事実は、キナーゼPAK1がイベルメクチンの新しい標的である可能性を示唆している。
このキナーゼは、膵臓がん、結腸がん、乳がん、前立腺がん、NF(神経線維腫症)腫瘍などのヒトのがんの70%以上の増殖に必要である。
この研究では、イベルメクチンがヒト卵巣がんおよびNF2欠損シュワン細胞腫細胞株の発がん性キナーゼPAK1を阻害し、細胞培養におけるPAK1依存性の増殖を抑制することを初めて実証した。50%増殖阻害濃度(IC50)は細胞株に応じて5〜20μMの間であった。
以下のような報告があります。フランスの研究グループからの論文です。
Macrocyclic lactones inhibit nasopharyngeal carcinoma cells proliferation through PAK1 inhibition and reduce in vivo tumor growth.(大環状ラクトンは、PAK1阻害を介して鼻咽頭がん細胞の増殖を阻害し、in vivoでの腫瘍成長を抑制する)Drug Des Devel Ther. 2018 Sep 7;12:2805-2814.
【要旨】
目的:エプスタイン-バーウイルス(Epstein-Barr virus :EBV)に関連するがんである鼻咽頭がんは、ヨーロッパおよび北米では稀であるが、南アジア、北アフリカ、イヌイットなど、世界の一部の地域では公衆衛生上の問題となっている。
鼻咽頭の解剖学的構造と位置のため、原発鼻咽頭がんの治療に手術が使用されることはほとんどない。化学療法と併用または非併用の放射線療法による治療は、原発腫瘍には有効であるが、致命的な再発や転移を防ぐことはできない。
方法:高容量スクリーニングによる新しい治療用分子の検索は、有望な薬剤としてのイベルメクチンを同定した。イベルメクチンは、米国食品医薬品局(FDA)が承認した大環状ラクトンであり、駆虫剤および殺虫剤として広く使用されており、がんに対する有効性も示されている。
結果:この研究では、イベルメクチンが鼻咽頭がん細胞に対してin vitroの実験系で細胞傷害活性を持ち、PAK-1活性の阻害によるMAPK経路の活性化と抑制することを示す。さらに、テストされたすべての大環状ラクトンとPAK1阻害剤は、EBV陽性およびEBV陰性の鼻咽頭がん細胞に対してin vitroで細胞毒性を示す。また、米国食品医薬品局(FDA)が承認した用量でのイベルメクチンの腹腔内反復注射は、有意な毒性がなく、ヌードマウスの皮下に移植した鼻咽頭がんの増殖を減少させることも示した。
結論:大環状ラクトンは、検出可能な有害作用なしにPAK-1を標的とする鼻咽頭がんに対する有望な治療薬と思われる。
以下のような報告があります。中国からの論文です。
Ivermectin Induces Cytostatic Autophagy by Blocking the PAK1/Akt Axis in Breast Cancer (イベルメクチンは乳がん細胞のPAK1 / Akt経路を遮断することにより細胞増殖抑制性オートファジーを誘導する)Cancer Res;2016 Aug 76(15); 4457–69.
【要旨】
乳がんは世界中の女性の間で最も一般的ながんであるが、治療の成功は依然として臨床的課題である。広範囲の抗寄生虫薬であるイベルメクチンは、多くの研究で抗腫瘍効果が示され、抗がん剤として可能性が報告されている。しかし、その抗がん作用に関する分子メカニズムはまだ十分に解明されていない。
ここでは、in vitroおよびin vivoの両方の実験系において、細胞増殖抑制性オートファジーを活性化することにより、乳がんを抑制するイベルメクチンの作用を報告する。
メカニズム的には、乳がん細胞におけるイベルメクチン誘発性のオートファジーは、ユビキチン化媒介分解経路を介したP21活性化キナーゼ1(PAK1)発現の減少と関連している。
PAK1の阻害は、Aktのリン酸化レベルを低下させ、Akt / mTORシグナル伝達経路を遮断する。乳がんの異種移植腫瘍の実験では、イベルメクチン誘導性の細胞増殖抑制性オートファジーにより、腫瘍の成長が抑制される。
以上の結果から、我々の実験結果は、イベルメクチンを使用して乳がん細胞の増殖を阻害する分子メカニズムを明らかにし、イベルメクチンが乳がん治療の潜在的な選択肢であることを示している。
オートファジー(自食作用)というのは、細胞内のタンパク質を分解してリサイクルする現象です。飢餓状態になったときに、自分の細胞を分解して栄養源にするのが本来の目的ですが、細胞内の異常タンパク質を除去する作用もあります。
がん細胞においては、ダメージを受けたタンパク質を分解して、再利用することによって細胞の生存を維持する効果があります。
このようにオートファジーは細胞を保護する役割がありますが、オートファジーが亢進すると細胞死が起こります。イベルメクチンは後者の細胞増殖抑制性オートファジーを誘導するという研究結果です。イベルメクチンはPAK-1のユビキチン介在性の分解を亢進し、PAK-1/Akt/mTOR経路を阻害してオートファジーを促進するというメカニズムです。
活性化したAktがシグナル伝達の下流に位置する様々なタンパク質をリン酸化して生存や増殖を制御しています。したがって、PAK-1阻害を介してAkt活性を抑制することはがん細胞の増殖抑制に重要なメカニズムです。

図:セリン/スレオニンキナーゼのAktは、多くのシグナル伝達経路のネットワークの中心的存在で、下流の幅広いターゲット分子や相互作用分子を介してさまざまな細胞内反応を引き起こす。PAK-1はAktやRaf/MEK/Erk経路を亢進してがん細胞の増殖や生存を促進する。
以上のように、イベルメクチンのPAK阻害作用は世界中のがん研究者が注目しています。がん治療のターゲットとしてのPAKの重要性と、その安全性の高さを考慮すると、PAK阻害剤としてのイベルメクチンの利用は極めて有用だと思います。

図:低分子量Gタンパク質のRASは細胞外のさまざまな刺激(チロシンキナーゼ受容体やサイトカイン受容体やカルシウムチャネルなど)を受けて細胞増殖や生存を促進するRAFキナーゼ(Raf-1)やPI-3キナーゼ(PI3K)など多数のシグナル伝達系を活性化する(①)。p21活性化キナーゼ(PAK-1)はRacおよびCdc42のような低分子量GTPaseによって活性化される(②)。PAK-1はRasシグナル伝達系の主要な経路であるRaf-1/MEK1/ERK経路(③)とPI3K/AKT経路(④)を活性化する。さらに、PAK-1はβ-カテニンをリン酸化してWNT/β-カテニン経路を活性化する(⑤)。このように、PAK-1は複数のシグナル伝達系の制御に関与し、がん細胞の増殖と転移を促進する(⑥)。寄生虫治療薬のイベルメクチン(Ivermectin)はPAK-1とWNT/β-カテニン経路を阻害する作用によって、抗腫瘍作用を発揮する(⑦)。
【カンナビジオールはPAK-1を阻害する】
以下のような報告があります。
Cannabinoids Inhibited Pancreatic Cancer via P-21 Activated Kinase 1 Mediated Pathway.(カンナビノイドは、P-21活性化キナーゼ1媒介経路を介して膵臓がんを阻害した)Int J Mol Sci. 2020 Nov; 21(21): 8035.
【要旨】
膵臓がんの場合、カンナビジオール(CBD)やΔ9-テトラヒドロカンナビノール(THC)などのカンナビノイドの抗がん効果が報告されている。膵臓がんの90%以上で変異しているがん遺伝子のKrasや、免疫チェックポイント阻害の主要な標的であるPD-L1に対するCBDやTHCの作用は、十分に検討されていない。
膵臓がんの細胞株とマウスの移植がんモデルを使用して、膵臓がん細胞の増殖や、膵臓がんと間質細胞(膵星細胞)に対するCBDとTHCの影響について検討した。CBDとTHCは、膵臓がん細胞、膵星細胞、および膵臓星状細胞で刺激された膵臓がん細胞の増殖を抑制した。それらはまた、マウスに移植した膵臓がんの成長を抑制した。
さらに、CBDおよび/またはTHCは、膵臓がん細胞または膵星細胞のいずれかによるPD-L1の発現を減少させた。
p-21活性化キナーゼ1(PAK1)の遺伝子ノックアウトマウスでは、CBDおよびTHCのこれらの効果は顕著に抑制された。
これらの結果は、CBDとTHCがp-21活性化キナーゼ1(PAK1)の阻害を介して膵臓がん細胞と膵星細胞に作用することを示唆している。PD-L1発現のCBDおよびTHCによる阻害は、膵臓がんに対する免疫チェックポイント阻害剤による治療効果を強化する。
以上から、イベルメクチンとカンナビジオールはPAK1阻害を介して、抗腫瘍免疫を増強し、免疫チェックポイント阻害剤の効き目を高める効果が期待できます。
| « 798)食事から... | 800) がん細胞... » |