がんの予防や治療における漢方治療の存在意義を考察しています。がん治療に役立つ情報も紹介しています。
「漢方がん治療」を考える
カレンダー
| 2025年1月 | ||||||||
| 日 | 月 | 火 | 水 | 木 | 金 | 土 | ||
| 1 | 2 | 3 | 4 | |||||
| 5 | 6 | 7 | 8 | 9 | 10 | 11 | ||
| 12 | 13 | 14 | 15 | 16 | 17 | 18 | ||
| 19 | 20 | 21 | 22 | 23 | 24 | 25 | ||
| 26 | 27 | 28 | 29 | 30 | 31 | |||
|
||||||||
goo ブログ
過去の記事
カテゴリ
最新の投稿
最新のコメント
最新のトラックバック
ブックマーク
プロフィール
検索
gooおすすめリンク



| URLをメールで送信する | |
| (for PC & MOBILE) | |
856)ケトン体療法(その6):SGLT2阻害剤と全体のまとめ
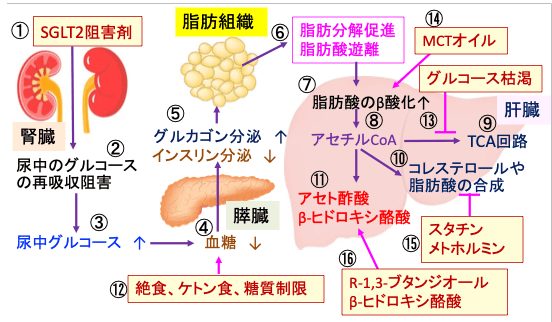
図:SGLT2(ナトリウム-グルコース共輸送体2)阻害剤(①)は、腎臓で濾過されたグルコースの再吸収を阻害し(②)、尿中にグルコースを排出(③)ことによって血糖を低下する(④)。その結果、膵臓からのインスリン分泌が低下し、グルカゴン分泌が増える(⑤)。このホルモン分泌の変化は脂肪組織の脂肪分解と脂肪酸の遊離を促進する(⑥)。脂肪酸は肝臓でβ酸化によって代謝され(⑦)、アセチルCoAが増える(⑧)。アセチルCoAはミトコンドリアのTCA回路で代謝され(⑨)、細胞質ではコレステロールや脂肪酸の合成に使われる(⑩)。グルコースの利用が制限された条件(絶食や糖質制限)では、アセチルCoAはケトン体(アセト酢酸とβ-ヒドロキシ酪酸)の合成に使われる(⑪)。絶食・ケトン食・糖質制限(⑫)は血糖を低下させ、グルコースを枯渇してTCA回路での代謝を抑制する(⑬)。MCTオイル(⑭)は脂肪酸のβ酸化を促進してアセチルCoAを増やす。スタチンとメトホルミンはコレステロール合成と脂肪酸合成を抑制してケトン体合成に使われるアセチルCoAを増やす(⑮)。R-1.3-ブタンジオールやβ-ヒドロキシ酪酸などの外来性ケトンの摂取はさらにケトン体を増やす(⑯)。これらを併用すると、ケトン体濃度を高めることができる。
856)ケトン体療法(その6):SGLT2阻害剤と全体のまとめ
【糖尿病治療薬には老化抑制と寿命延長に有効な薬がある】
糖尿病はインスリンの分泌量が低下したり、インスリンの効き目が低下して、血糖が上昇する病気です。血中のグルコース濃度が高くなるので、様々な組織や臓器にダメージをあたえて、老化を促進し、寿命を短くします。
インスリンを投与して血糖を下げれば、グルコース(ブドウ糖)による組織障害は減少しますが、インスリンは様々なメカニズムで老化を促進し、寿命を短くします。インスリンはがん細胞の増殖を促進します。
つまり、血糖を低下させる効果があっても、インスリンは老化とがん細胞の発生を促進し、寿命を短縮します。
一方、インスリンの血中濃度を高めない糖尿病治療薬は、老化抑制と寿命を延ばす効果が報告されています。抗老化薬の探索に関する最近の総説で以下のような論文があります。
Anti-ageing effects of FDA-approved medicines: a focused review(FDA承認薬の老化防止効果:焦点を絞ったレビュー)J Basic Clin Physiol Pharmacol. 2023 Jan 13;34(3):277-289.
【要旨の抜粋】
老化は、生活の質に影響を与える加齢関連疾患の原因となる。人々は抗老化(アンチエイジング)の方法に興味を持っており、多くの科学者がアンチエイジング薬の探索を試みている。
この論文で米国食品医薬品局(FDA)が承認した医薬品の老化防止活性を調べ、アログリプチン(alogliptin)、カナグリフロジン(canagliflozin)、メトホルミン(metformin)が AMPK 活性化を介して老化防止活性を生み出す可能性があることを発見した。
ラパマイシン(rapamycin)とカナグリフロジン(canagliflozin)はmTORを阻害して寿命を延ばすことができる。アトラクリウム(atracurium)、カルニチン(carnitin)、スタチン(statin)は DAF-16 活性化因子として作用し、抗老化活性に寄与する可能性がある。
アカルボース(acarbose)はインスリン低下効果によって長寿を促進する可能性がある。
興味深いことに、一部の薬物 (カナグリフロジン、メトホルミン、ラパマイシン、アカルボースなど) は主に雄の動物で寿命延長効果を示す可能性がある。
この論文の中でアログリプチン(alogliptin)、カナグリフロジン(canagliflozin)、メトホルミン(metformin)、アカルボース(acarbose)が糖尿病の治療薬として使用されています。
寿命を延ばす方法として現時点で最も確実なのがカロリー制限です。カロリー制限とは、栄養障害(ビタミンやミネラルやタンパク質の不足)を起こさずに食事からの摂取カロリーを30~40%程度減らす食事を行うことです。カロリー制限には老化を遅延して寿命を延ばし、がんを含めて老化関連疾患の発症を抑制する効果が認められています。
細胞の増殖と老化の制御で重要な役割を担っているのがmTORC1(哺乳類ラパマイシン標的タンパク質複合体1)です。
男性が女性より寿命が短いのは、このmTORC1活性が男性の方が高いという考えもあります。男性は屈強さを得るために寿命を犠牲にしているという考えです。
前述の論文で『一部の薬物 (カナグリフロジン、メトホルミン、ラパマイシン、アカルボースなど) は主に雄の動物で寿命延長効果を示す可能性があります。』というのは、これらの薬がmTORC1活性の抑制に関係している可能性を示唆しています。
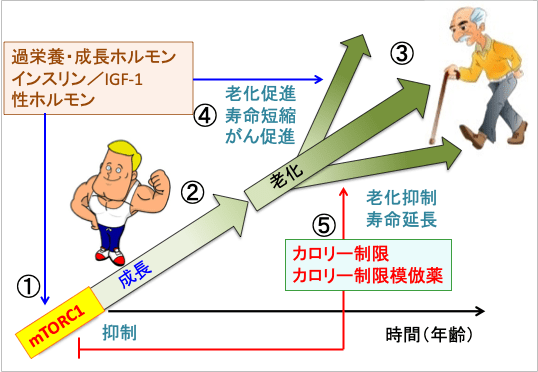
図: mTORC1(哺乳類ラパマイシン標的タンパク質複合体1)は成長ホルモンやインスリンやインスリン様成長因子-1(IGF-1)など様々な成長因子や過剰な栄養によって活性化され(①)、細胞の増殖や体の成長を促進する役割を担っている(②)。成長が終了したあともmTORC1の働きが過剰に続くと、細胞や組織の老化が促進される(③)。成長は「プログラムされた正常機能」であるが、老化は「成長の延長(過剰機能)」であり、成長終了後はmTORC1の活性は老化と発がんを促進する方向に作用する(④)。mTORC1を活性化して屈強な体を作るときは、寿命を犠牲にし、発がんリスクを高める可能性がある。カロリー制限とカロリー制限模倣薬はmTORC1の活性を抑制することによって、老化速度を遅くし、寿命を延長できる(⑤)。
カロリー制限と同じ効果(抗老化や寿命延長効果)を示す薬をカロリー制限模倣化合物(Calorie restriction mimetics :CRM)と言います。カロリー制限模倣化合物には抗糖尿病薬のメトホルミン、赤ワインに含まれるレスベラトロール、ポリアミンの一種のスペルミジンなどが知られています。
メトホルミンは、ミトコンドリアの呼吸鎖の最初のステップである呼吸酵素複合体I を阻害します。その結果、ミトコンドリアでのATP産生が減少し、AMP:ATPの比が上昇し、AMP活性化プロテインキナーゼ(AMPK)が活性化されます。活性化したAMPKは、肝臓の糖新生を抑制し、解糖を亢進し、骨格筋でのグルコース利用を促進して血糖を低下させます。
AMPKはmTORC1を阻害します。mTORC1は老化と発がん過程の両方を促進する働きがあるので、mTORC1の抑制は抗老化と抗がんの両方の効果になります。

図:メトホルミンは絶食やカロリー制限や運動と同様に、体内のエネルギー低下によってAMP/ATP比とNAD+/NADH比を高める(①)。AMP/ATP比の上昇はAMP活性化プロテインキナーゼ(AMPK)を活性化し(②)、NAD+/NADH比の上昇はサーチュイン1(Sirtuin 1)を活性化する(③)。サーチュイン1はセリン・スレオニン・キナーゼのLKB1を活性化し、LKB1はAMPKを活性化する(④)。LKB1はAMPKを活性化し(⑤)、AMPKはサーチュイン1を活性化する(⑥)。サーチュイン1はPGC-1αの活性を亢進する(⑦)。PGC-1αは、ミトコンドリア新生を亢進して数を増やし、脂肪酸β酸化やTCA回路や酸化的リン酸化などミトコンドリア機能を亢進する(⑧)。その結果、抗老化と寿命延長の効果を発揮する。
アカルボースは糖を分解する酵素の働きを阻害して、体内へのブドウ糖の吸収を減らす薬です。血糖の上昇とインスリンの分泌を抑制して、抗老化と寿命延長効果を発揮します。アカルボースの寿命延長効果は831話で解説しています。

図:食事中の糖質(①)は唾液や膵液のα-アミラーゼで二糖類(②)になり、小腸のα-グルコシダーゼによって単糖類(③)となって腸から吸収される。血糖とインスリンの上昇は老化を促進し、寿命を短縮し、がん細胞の発生と増殖を促進する(④)。アカルボースはα-アミラーゼとα-グルコシダーゼの両方を阻害する(⑤)。糖質と二糖類は腸から吸収できないので、腸内細菌によって分解され、酪酸などの短鎖脂肪酸が増える(⑥)。短鎖脂肪酸は老化を抑制し、寿命を延ばし、がん細胞の発生と増殖を抑制する(⑦)
【SGLT2(ナトリウム-グルコース共輸送体2)阻害剤は尿にグルコースを排出する】
腎臓は体内の老廃物を尿中に排泄する働きがあります。まず、腎臓の糸球体で血液中の物質(水、塩類、有機物などの小さな分子)が濾過されます。濾過されたばかりの尿を原尿と言い、
この原尿から、体に有用な物質を再び血液中に戻すために尿細管で再吸収が行われます。(下図)

図:腎臓の糸球体で濾過された原尿中のグルコースは尿細管でほとんどが再吸収されている。
グルコースは、栄養素として非常に重要な糖であるため、通常は原尿中のグルコースの99%以上が再吸収されるので、血糖値と腎臓が正常な人は尿にはグルコースはほとんど含まれません。通常、健康な人の尿細管でのグルコース再吸収量は、1日におおよそ180グラムから200グラムです。
しかし、糖尿病になって血糖値が上昇すると、濾過されたグルコースを全て再吸収できないので、尿糖が出るようになります。これが糖尿病です。
尿細管で尿中のグルコースを再吸収するのがSGLT2というタンパク質です。
SGLT2は、Sodium-Glucose Cotransporter 2(ナトリウム-グルコース共輸送体2)の略です。SGLT2の働きを阻害するSGLT2阻害薬は、2型糖尿病の治療に使用されています。
SGLT2を阻害することで、腎臓が尿中に余分なグルコースを排出するよう促す効果があり、これにより血糖値を下げる効果があります。さらにインスリン分泌も低下するので、老化を抑制し、寿命を延ばすことになります。(下図)

図:SGLT2(ナトリウム-グルコース共輸送体2)阻害剤(①)は、腎臓で濾過されたグルコースの再吸収を阻害し(②)、尿中にグルコースを排出(③)ことによって血糖を低下する(④)。その結果、インスリン分泌が低下する(⑤)。血糖とインスリンの低下は抗老化と寿命延長効果となる。
【SGLT2 阻害剤は心臓病や腎臓病による死亡率を低下する】
SGLT2 阻害剤には、血糖降下効果に加えて、腎臓と心臓に対して広範囲の有益な作用(腎臓保護作用、心不全の発症予防など)が報告されています。例えば、以下のような報告があります。
SGLT-2 inhibitors in patients with heart failure: a comprehensive meta-analysis of five randomised controlled trials(心不全患者におけるSGLT-2阻害剤:5つのランダム化対照試験の包括的なメタ分析)Lancet. 2022 Sep 3;400(10354):757-767.
複数の臨床試験のメタ解析の結果、心不全患者にSGLT-2阻害剤を使うと、心血管死が87%、全死因死亡が92%に減少することが明らかになりました。この論文の結論は『SGLT2 阻害剤は、幅広い心不全患者の心血管死および心不全による入院のリスクを軽減し、駆出率や治療環境に関係なく、心不全の基礎療法として有効』となっています。
以下のような総説論文もあります。
Roles for SGLT2 Inhibitors in Cardiorenal Disease(心腎疾患におけるSGLT2阻害剤の役割)Cardiorenal Med. 2022;12(3):81-93.
SGLT2阻害剤を投与された慢性腎臓病の患者は、プラセボを投与された患者よりも腎機能の持続的低下、末期腎疾患、または腎または心血管疾患による死亡のリスクが低下しました。
SGLT2阻害剤を投与された心疾患患者では、心不全による入院や死亡のリスクが、プラセボ群に比べて有意に低下しました。
SGLT2阻害剤は、2型糖尿病の存在や、慢性腎疾患または心疾患の重症度に関係なく、病気の進行を抑えて、死亡リスクを低下させる効果が観察されました。
FDA承認薬から老化防止効果のある薬を探索する研究の総説(一番最初の論文)では、SGLT2阻害剤のカナグリフロジン(canagliflozin)が、『AMPK 活性化を介して老化防止活性を生み出す可能性』と『mTORを阻害して寿命を延ばす可能性』を指摘しています。
カナグリフロジンは『カナグル』という商品名で糖尿病治療に使われていますが、他のSGLT2阻害剤も同様な効果が期待できます。
尿に糖を排泄するというのは、食料の無駄になり、糖尿による尿路感染症のリスクもあるのですが、抗老化と寿命延長に効果が期待できるようです。最近の研究では、SGLT2阻害剤が体内のケトン体を増やす作用があり、このケトン体増加が抗老化と寿命延長のメカニズムに関与していることが指摘されています。
さらに、ケトン体産生とは関係ないメカニズムでがん細胞の増殖を抑える作用が報告されています。これに関しては次回解説します。
【SGLT2阻害剤はリンゴの木の樹皮から見つかった】
1835年、フランスの化学者が、リンゴの木の樹皮からフロリジンと呼ばれる分子を単離しました。(下図)

図:フロリジン(phlorizin)はリンゴの木の幹の樹皮から見つかった。
発見された当時、フロリジンは発熱、感染症、マラリアの治療薬候補であると考えられていました。
1886年にはフロリジンが尿糖を誘発することが明らかになり、1903年にはフロリジンが動物の空腹時と食後の高血糖を正常化し、糖尿病の治療薬となる可能性があることが発見されました。
フロリジンが腎臓の近位尿細管刷子縁におけるグルコースの再吸収を担うトランスポーターに作用することは、1970年代初頭に明らかになりました。 1980 年代後半から 1990 年代にかけて、実際に関与するグルコーストランスポータータンパク質は SGLT タンパク質として同定されました。
フロリジンは動物実験では高血糖とインスリンレベルを正常化しましたが、ヒトの糖尿病治療薬としては不適格でした。その理由は、フロリジンは胃腸管からの吸収(経口バイオアベイラビリティ)が低く、第二に、フロリジンは SGLT1 (主に胃腸管に存在) と SGLT2 の両方を阻害し、副作用(下痢などの消化器症状)のを引き起こします。 第三に、フロレチン (フロリジンの代謝産物) は、さまざまな組織でのグルコースの取り込みを制限する重要なグルコース トランスポーター 1 (GLUT1) を強力に阻害します。
その後、これらの問題を解決するフロリジンの類似体が開発されています。経口で生物学的に利用可能な選択的 SGLT2 阻害剤の発見競争は1990 年代に始まりました。
2013年3月に田辺三菱のカナグリフロジンがFDAによって承認された最初のSGLT2阻害剤となり、続いて2014年1月にヤンセンのダパグリフロジン、2014年8月にベーリンガーインゲルハイムのエンパグリフロジンが続きました。
現在糖尿病患者に使用されている実用的なSGLT2阻害剤は全てフロリジン誘導体となっており、まさにフロリジンはSGLT2阻害剤の元祖ともいえる存在です。
【SGLT2阻害剤はケトーシス(ケトン血症)を引き起こす】
ケトン体は、19 世紀半ばに糖尿病性ケトアシドーシスで亡くなった患者の尿中から初めて大量に発見されたため、当時の医師はケトン体をグルコース代謝障害の有毒な副産物と見なしていました。
医学者が、糖質と糖原性アミノ酸の供給源が不足している場合、ケトン体が肝臓によって生成される正常な代謝産物であり、その量が増加することを理解するのにほぼ半世紀かかりました。残念なことに、断食中またはケトン食を実践している間に健康な人に発生する安全な「生理的なケトン血症」と、インスリン欠乏性糖尿病に関連する病的で制御不能な高ケトン血症とを区別できない医師もまだいます。
伝統的に、医師はケトーシスを恐れるように教えられてきました。これは、インスリン欠乏に起因する顕著な高ケトン血症が重度のアシドーシス(酸性血症)と 1 型糖尿病患者の死亡を引き起こす可能性があるためです。
しかし、ケトン体が増えるケトーシスはがん治療だけでなく、認知症などの神経変性疾患、循環器疾患の治療に有効であることが明らかになっています。ケトン体の抗老化作用や寿命延長効果も明らかになり、アンチエイジングの領域でも注目されています。
したがって、ある薬にケトン体合成を誘発あるいは促進する薬があると、その薬によって起こるケトーシスは「副作用」と捉えられる可能性があります。しかし、ケトン体を増やすという作用が病気の治療に利用する発想もあります。ケトン体を増やすケトンサプリメントが、老化や認知症やがんや心血管疾患の治療に利用されているのと同じです。
脂肪酸合成を阻害するメトホルミンやメバロン酸経路を阻害するスタチンはアセチルCoAをケトン体合成に回すことによって内因性ケトン合成を促進します。
PPARαを活性化するフェノフィブラートやベザフィブラートは、HMG-CoAからアセト酢酸を生成するHMG-CoAリアーゼの発現を誘導することによってケトン体合成を亢進します。
つまり、これらを併用すると内因性のケトン体合成を促進できます。(853話参照)
SGLT2阻害剤は脂肪酸の分解(β酸化)を促進し、ケトン体合成を亢進します。
以下のような報告があります。
The SGLT2 inhibitor dapagliflozin promotes systemic FFA mobilization, enhances hepatic β-oxidation, and induces ketosis(SGLT2 阻害剤ダパグリフロジンは、全身の FFA 動員を促進し、肝臓のβ酸化を促進し、ケトーシスを誘導する)J Lipid Res. 2022 Mar; 63(3): 100176.
SGLT2阻害剤は尿糖を増やして血糖を低下させるので、インスリンが低下し、グルカゴンが増えます。その結果、脂肪組織からの脂肪酸の動員と肝臓における脂肪酸のβ酸化が亢進し、できたアセチルCoAはグルコースが枯渇した条件ではケトン体合成に振り向けられます。(下図)

図:SGLT2(ナトリウム-グルコース共輸送体2)阻害剤(①)は、腎臓で濾過されたグルコースの再吸収を阻害し(②)、尿中にグルコースを排出(③)ことによって血糖を低下する(④)。その結果、膵臓からのインスリン分泌が低下し、グルカゴン分泌が増える(⑤)。このホルモン分泌の変化は脂肪組織の脂肪分解と脂肪酸の遊離を促進する(⑥)。脂肪酸は肝臓でβ酸化によって代謝され(⑦)、アセチルCoAが増える(⑧)。アセチルCoAはミトコンドリアでTCA回路で代謝され(⑨)、コレステロールや脂肪酸の合成に使われる(⑩)。グルコースの利用が制限された条件(絶食や糖質制限)では、アセチルCoAはケトン体(アセト酢酸とβ-ヒドロキシ酪酸)の合成に使われる(⑪)。
SGLT2阻害剤によって誘発される尿中グルコース排泄が、エネルギー源としてグルコースの代わりに脂肪酸の使用を促進することにより、安定したカロリー損失を伴い脂肪量を特異的に減少させることを示しています。高血糖を改善し、体重減少を促進することにより、SGLT2阻害剤は肥満者の 2 型糖尿病の治療に有用であることが示されています。
脂肪酸分解の促進によってケトン体合成が増えます。この現象は、副作用の原因になる可能性がありますが、ケトン体の様々な健康作用の観点から有用な効果のメカニズムという考えも出てきます。
【SGLT2阻害剤によるケトーシス(ケトン血症)誘導は副作用の原因になる】
前述の作用機序からSGLT2阻害剤を服用して絶食すると、脂肪酸の燃焼が更新してケトン体産生が増えます。
通常の食事をしながらSGLT2阻害剤を服用しても、血糖上昇が抑えられ、インスリン分泌も低下しますが、ケトーシスは通常起こりません。
しかし、絶食やインスリン作用が低下した場合は、ケトン体産生を増え、ケトーシスが起こります。たとえば、糖尿病患者でインスリンの量が極端に低下した状態ではケトアシドーシスが起こる可能性があります。以下のような論文があります。
SGLT2 Inhibitors May Predispose to Ketoacidosis.(SGLT2阻害剤はケトアシドーシスの素因となる可能性がある)J Clin Endocrinol Metab. 2015 Aug; 100(8): 2849–2852.
【要旨の抜粋】
ナトリウム・グルコース共輸送体 2 (SGLT2) 阻害剤は、ブドウ糖の尿中排泄を増加させ、それによって血糖コントロールを改善し、体重減少を促進する抗糖尿病薬である。
2013 年にこのタイプの薬が承認されて以来、これらの薬が糖尿病性ケトアシドーシスのリスクを高めることを示唆するデータが明らかになった。2015年5月、食品医薬品局は、SGLT2阻害剤がケトアシドーシスを引き起こす可能性があると警告を発した。
SGLT2 阻害剤は、糖尿病性ケトアシドーシスの素因となる可能性のある複数のメカニズムを引き起こす。SGLT2 阻害剤をインスリンと組み合わせる場合、低血糖を避けるためにインスリンの用量を減らす必要がある。より低い用量のインスリンは、脂肪分解およびケトン生成を抑制するには不十分である。さらに、SGLT2は膵臓α細胞で発現しており、SGLT2阻害剤はグルカゴンの分泌を促進する。
最後に、SGLT ファミリートランスポーターの非選択的阻害剤であるフロリジンは、ケトン体の尿中排泄を減少させる。ケトン体の腎臓クリアランスの減少も、血漿ケトン体レベルを増加させる可能性がある。
この論文は2015年8月の報告です。2013年3月に田辺三菱のカナグリフロジンが米国の食品医薬品局 (FDA) から承認されて2年後の2015 年 5 月に食品医薬品局 (FDA) が SGLT2 阻害剤による治療によりケトアシドーシスのリスクが増加する可能性があると警告しています。
FDAの警告には、SGTL2阻害剤が1型糖尿病と2型糖尿病の両方の患者のケトアシドーシスのリスクを高めることを示唆する医学文献の報告が先行しました。
例えば、1型糖尿病患者を対象とした8週間の研究では、エンパグリフロジンで治療を受けた患者の約5%(42人中2人)が糖尿病性ケトアシドーシスを発症したため研究から離脱しました。
さらに、SGLT2阻害剤は、負に帯電したケトン体の再吸収を促進することが指摘されています。
つまり、SGLT2阻害剤はケトン体生成の増加と腎クリアランスの減少の組み合わせにより、循環ケトン体レベルを増加させる相加効果を発揮します。したがって、インスリン依存性の 1型糖尿病患者に SGLT2 阻害剤を投与すると、循環ケトン体レベルが上昇し、ケトアシドーシスを発症しやすくなるというのは生物学的に考えられます。
最近の論文では、SGLT2 が膵臓のα細胞で発現していることが報告されました。そして、このトランスポーターはα細胞のグルコースセンサー機構の一部として機能しているようです。さらに、ヒト膵島をSGLT2阻害剤のダパグリフロジンに曝露すると、おそらくα細胞機能に直接影響を及ぼしてグルカゴン分泌が増加しました。グルカゴン分泌を促進する SGLT2 阻害剤の作用は、ケトン体生成を増加させる強力な推進力を提供すると考えられます。
さらに、動物研究では、グルカゴンが肝臓に作用してキスペプチン-1の分泌を促進し、その結果、グルコース刺激によるインスリン分泌が抑制されることが実証されました。このようなメカニズムがヒトで機能している場合、一部の患者では内因性インスリン分泌が減少し、ケト生成がさらに促進される可能性があります。
【SGLT2阻害剤は尿からのケトン体再吸収を促進する】
血中のケトン体が増えると尿中のケトン体濃度も増えます。ケトン体はグルコースの代わりになるエネルギー源なので、生体に有用な成分です。そのため、尿中に濾過された原尿中のケトン体は尿細管で再吸収されます。以下のような報告があります。
Tubular reabsorption and urinary excretion of acetoacetate and 3-hydroxybutyrate in normal subjects and juvenile diabetics(健常者および若年性糖尿病患者におけるアセト酢酸および3-ヒドロキシ酪酸の尿細管再吸収と尿中排泄)Acta Med Scand. 1977 Jan;201(1-2):63-7.
アセト酢酸と 3-ヒドロキシ酪酸の腎臓での取り扱いについて、健常者 8 名とインスリン治療を受けた若年糖尿病患者 7 名を対象に、DL-3-ヒドロキシ酪酸ナトリウムの静注前後で検査されました。
健常者でも糖尿病患者でもケトン体は再吸収されました。アセト酢酸および 3-ヒドロキシ酪酸の濾過速度が低い場合、再吸収はほぼ完全でした。
濾過速度の増加に伴い、アセト酢酸および 3-ヒドロキシ酪酸の尿細管再吸収速度と尿中排泄速度の両方が直線的に増加しました。糖尿病患者ではアセト酢酸および 3-ヒドロキシ酪酸の濾過速度が高いにもかかわらず、どちらのケトン体の平均再吸収率も正常者で見られるものと同じでした。
3-ヒドロキシ酪酸はβヒドロキシ酪酸と同じです。ラセミ体のDL-3-ヒドロキシ酪酸ナトリウムを静注して、尿中の排泄と再吸収を検討すると、ケトン体は糸球体で濾過されたあと、尿細管で再吸収されています。ケトン体の濾過量が少なければ完全に再吸収されますが、濾過されるケトン体が増えると、それに比例して、再吸収量と尿中への排泄量が増えるということです。
血糖が上がると尿糖が増えるのと同じで、ケトン体も血中濃度が増えると、再吸収速度に限界があるので、尿中に排泄されるケトン体も増えます。以下のような論文もあります。
Renal conservation of ketone bodies during starvation(飢餓時の腎臓のケトン体の保存)Metabolism. 1975 Jan;24(1):23-33.
完全な絶食状態にある12人の肥満被験者を対象に、アセト酢酸とβ-ヒドロキシ酪酸の腎臓での処理が研究されました。アセト酢酸、β-ヒドロキシ酪酸、およびイヌリンのクリアランス速度が測定され、アセト酢酸およびβ-ヒドロキシ酪酸の再吸収速度が計算されました。
絶食期間中、血中アセト酢酸およびβ-ヒドロキシ酪酸の腎臓クリアランス速度は一定のままでした。対照的に、アセト酢酸塩の再吸収速度は、3 日目の 47 ± 10 μモル/分から、10、17、および 24 日目には 106 ± 15、89 ± 10、および 96 ± 10 μモル/分へと大幅に増加しました。
同様に、β-ヒドロキシ酪酸の再吸収速度は、3 日目の 154 ± 27 μモル/分から、10 日目、17 日目、および 24 日目には 419 ± 53 μモル/分、399 ± 25 μモル/分、および 436 ± 53 μモル/分へと大幅に増加しました。
アセト酢酸とβ-ヒドロキシ酪酸の再吸収速度は、負荷に対して直線的に増加しました。
絶食期間に応じてケトン体の産生が増えますが、10日以上になると血中のケトン体濃度はほぼ一定になります。ケトン体の血中濃度が増え、尿中に濾過されるケトン体の量が増えると、再吸収速度も比例して増えます。
陰イオンのケトン体の排泄を減らせると、長期にわたる飢餓時の尿中からの陽イオンの大量損失が防止されます。アンモニウムは長期の絶食中に排泄される主要な陽イオンとなるため、ケトン体の腎臓による再吸収の増加により体タンパク質の損失が最小限に抑えられ、高い循環アセト酢酸濃度とベータヒドロキシ酪酸濃度の維持に役立ちます。
【SGLT2阻害剤によるケトーシス(ケトン血症)亢進は諸臓器を保護する】
糖尿病は、1 型糖尿病と呼ばれるインスリン欠乏症、または 2 型糖尿病と呼ばれるインスリン作用に対する抵抗性のいずれかに関連する慢性代謝疾患です。2型糖尿病は成人でより一般的であり、すべての糖尿病症例の約 90% を占めています。
ナトリウム・グルコース共輸送体 2 (SGLT2) 阻害剤は、最新のクラスの血糖降下薬の 1 つであり、現在、2型糖尿病の治療に強く推奨されています。
SGLT2 阻害剤による糖尿は通常、体重の減少をもたらし、それに伴う浸透圧利尿作用により最終的には血圧が低下します。したがって、SGLT2阻害剤は肥満や過体重の患者、および2型糖尿病を伴う高血圧において非常に有益です。さらに、SGLT2阻害剤は心血管機能と腎機能を良くする作用があります。
SGLT2阻害剤は低血糖を引き起こす傾向は低いですが、それでも、糖尿のため、生殖器感染症および尿路感染症の可能性があり、軽度の脱水症状により時折起立性低血圧を引き起こす可能性があります。さらに、前述のように米国食品医薬品局 (FDA) は 2015 年 5 月、1型糖尿病患者と2型糖尿病患者の両方におけるSGLT2阻害剤の使用に関連して糖尿病性ケトアシドーシス のリスクが増加する可能性があるという警告を発表しました。
しかし、最近の研究で、SGLT2阻害剤による心血管機能と腎機能の改善効果は、ケトン体産生が関与している可能性が指摘されています。例えば、以下のような報告があります。
SGLT2 Inhibition Mediates Protection from Diabetic Kidney Disease by Promoting Ketone Body-Induced mTORC1 Inhibition(SGLT2阻害はケトン体誘導性mTORC1阻害を促進することにより糖尿病性腎疾患からの保護を仲介する)Cell Metab. 2020 Sep 1;32(3):404-419.e6.
【要旨】
SGLT2 阻害剤は、糖尿病性腎疾患の患者に強力な腎保護をもたらす。 しかし、そのような保護作用のメカニズムは明らかではない。 今回我々は、非タンパク尿性糖尿病性腎疾患の動物モデル(高脂肪食を与えられたApoEノックアウトマウス)の損傷した近位尿細管において、哺乳類ラパマイシン標的タンパク質複合体1(mTORC1)の過剰活性化によりATP生成が脂肪分解依存からケトン体分解依存に移行したことを報告する。
さらに、SGLT2阻害剤のエンパグリフロジンが内因性ケトン体レベルを上昇させ、エンパグリフロジンの使用またはケトン体前駆体である1,3-ブタンジオールによる治療により、マウスの腎臓のATPレベルの低下と臓器損傷が防止されることも発見した。
エンパグリフロジンの腎保護効果は、ケトン体生成の律速酵素である Hmgcs2 (3-hydroxy-3-methylglutaryl-CoA synthase 2:HMG-CoA合成酵素2)の遺伝子欠失によって消失した。
さらに、ケトン体は糖尿病 db/db マウスにおける mTORC1 関連の糸球体上皮細胞の損傷とタンパク尿を軽減した。
我々の発見は、SGLT2阻害に関連する腎保護がケトン体の上昇によって媒介され、それが非タンパク尿性糖尿病性腎疾患およびタンパク尿性糖尿病性腎疾患で起こるmTORC1の過剰活性化を修正することを示している。
SGLT2阻害剤は、カロリー制限と同様に、燃料消費をグルコースから脂肪酸にシフトすることでケトン体濃度を増加させます。 SGLT2i阻害剤を介した糖尿病腎疾患からの保護には、ケトン体による mTOR 阻害が関与していることを指摘しています。
私の場合、SGLT2阻害剤を服用して1日絶食すると、6時間後以降βヒドロキシ酪酸の濃度は0.6mM から1.0mM程度まで上昇します。(下図)

図:朝食を抜いてSGLT2阻害剤のカナグリフロジンを100mg服用して、その後絶食すると血中のβ-ヒドロキシ酪酸の濃度は6時間後以降0.6mMを超え、12時間後には1.0mMに達する。
個人差はありますが、通常の絶食では、血中のβヒドロキシ酪酸濃度は12時間〜24時間の絶食で0.3mM〜0.5 mM、2〜3日の絶食で1.5 mM 、8日間の絶食で5 mM程度と言われています。SGLT2阻害剤を使うと絶食時のケトン体合成を促進する効果があります。
がん治療や抗老化の目的でケトン食やケトン体サプリメントで血中のケトン体を増やすとき、SGLT2阻害剤を利用すると、楽にケトン体を上げられます。ただし、副作用も知って、自己責任での使用になります。
| « 855)ケトン体... | 857)SGLT2阻害... » |