がんの予防や治療における漢方治療の存在意義を考察しています。がん治療に役立つ情報も紹介しています。
「漢方がん治療」を考える
カレンダー
| 2025年1月 | ||||||||
| 日 | 月 | 火 | 水 | 木 | 金 | 土 | ||
| 1 | 2 | 3 | 4 | |||||
| 5 | 6 | 7 | 8 | 9 | 10 | 11 | ||
| 12 | 13 | 14 | 15 | 16 | 17 | 18 | ||
| 19 | 20 | 21 | 22 | 23 | 24 | 25 | ||
| 26 | 27 | 28 | 29 | 30 | 31 | |||
|
||||||||
goo ブログ
過去の記事
カテゴリ
最新の投稿
最新のコメント
最新のトラックバック
ブックマーク
プロフィール
検索
gooおすすめリンク



| URLをメールで送信する | |
| (for PC & MOBILE) | |
747)FoxO3aを活性化すると膠芽腫(グリオブラストーマ)の幹細胞が消滅する
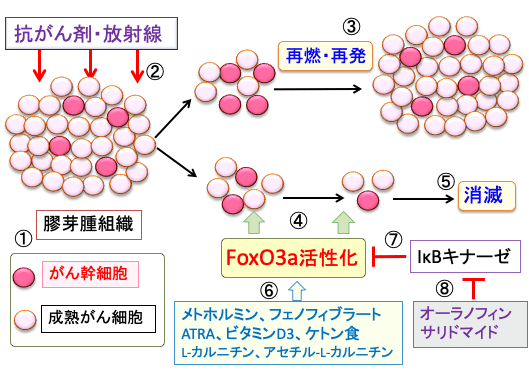
図:膠芽腫(グリオブラストーマ)はがん幹細胞と成熟がん細胞から構成されている(①)。抗がん剤治療や放射線治療(②)を行うと、成熟がん細胞は死滅するががん幹細胞は治療に抵抗性して生き残り、再燃・再発を引き起こす(③)。がん幹細胞のFoxO3aという転写因子を活性化すると(④)、がん幹細胞の特性を維持できなくなり、がん幹細胞の抗がん剤や放射線治療に対する感受性が高まり、がんを消滅できる(⑤)。メトホルミン、フェノフィブラート、ATRA(オールトランス・レチノイン酸)、ビタミンD3、ケトン食、L-カルニチン、アセチル-L-カルニチンはFoxO3aを活性化する(⑥)。IκBキナーゼはFoxO3aの転写活性を阻害し(⑦)、オーラノフィンとサリドマイドはIκBキナーゼ活性を阻害する作用によってFoxO3aを活性化する。これらを組み合わせると、FoxO3aを活性化して、膠芽腫を消滅できる。これらは、いずれも副作用はほとんどない。
747)FoxO3aを活性化すると膠芽腫(グリオブラストーマ)の幹細胞が消滅する
【転写因子FOXO3aを活性化すると膠芽腫のがん幹細胞が死滅する】
国立がんセンターと山形大学医学部の共同研究チームは、膠芽腫(グリオブラストーマ)のがん幹細胞の抗がん剤感受性を高める方法としてFoxO3aという転写因子の活性化が有効であることを2011年9月に報告しています。この研究で、FoxO3aを活性化する薬として経口糖尿病薬のメトホルミンの有効性を指摘しています。
膠芽腫の再発は、腫瘍組織中にごくわずかに存在するがん幹細胞(グリオブラストーマ幹細胞)が根源となることが分かっています。残りの大部分を占めるがん細胞は腫瘍を形成する能力を持たないため、取り残しても再発の恐れはないと考えられ、さらにがん幹細胞と比べると放射線や抗がん剤などの治療が効きやすいことも報告されています。つまり、このがん幹細胞を消滅することが膠芽腫の治療を成功させるポイントとなっています。
この研究では、膠芽腫の中に存在するがん幹細胞を腫瘍形成能力を持たないその他大勢のがん細胞に変える遺伝子としてFoxO3aを同定しています(下記の文献)。
FoxO3a functions as a key integrator of cellular signals that control glioblastoma stem-like cell differentiation and tumorigenicity.(FoxO3aは膠芽腫幹細胞の分化と腫瘍形成性をコントロールする細胞内シグナルの主要な調節器として作用する)Stem Cells. 29(9):1327-37. 2011年
そして、FoxO3aを活性化できる薬剤を探索すると、糖尿病治療薬のメトホルミンに膠芽腫幹細胞のFoxO3aを活性化する作用を見つけています(下記の文献)。
Glioma-initiating cell elimination by metformin activation of FOXO3 via AMPK.(メトホルミンによるAMPKを介したFOXO3の活性化によるグリオーマ始原細胞の排除)Stem Cells Transl Med. 1(11):811-24. 2012年
膠芽腫細胞を移植したマウスに、10日間メトホルミンを投与したところ、新たな腫瘍が形成されることなく、がん幹細胞が消失する効果が確認できたと報告しています。
つまり、膠芽腫幹細胞の幹細胞特性をメトホルミンが阻害し、がん幹細胞を腫瘍形成能力を持たないその他大勢の成熟がん細胞に変えることができるということです。腫瘍形成能力を喪失したがん細胞はそのうち自然消滅します。
【FoxO3aは転写因子】
FoxOは「Forkhead box O」の略で、DNA結合ドメインFOX(Forkhead box)をもつForkheadファミリーのサブグループ“O”に属する転写因子です。
哺乳類では4種類(FoxO1, FoxO3a, FoxO4, FoxO6)あり、線虫ではDaf-16、ショウジョウバエではdFOXOとそれぞれ1種類のみ存在します。
線虫とハエと哺乳類において、インスリン/インスリン様成長因子-1(IGF-1)シグナル伝達系は保存されており、FoxO転写因子はこのシグナル伝達系の下流する位置しています。
FoxO1とFoxO3aは約650個のアミノ酸からなる蛋白質で、遺伝子のプロモーター領域のTTGTTTACという配列に結合します。
転写因子というのは特定の遺伝子の発現(DNAの情報を蛋白質に変換すること)を調節している蛋白質で、FoxOはストレス応答、代謝制御、細胞周期、アポトーシス、細胞分化、DNA修復、免疫機能、炎症などに関連する多くの遺伝子の発現を促します。がん抑制遺伝子としての性格ももっており、FoxOの活性化は一般的に抗がん作用があります。
FoxO3aのリン酸化にはリン酸化されるセリンあるいはスレオニンの部位によって、核外に移行して転写活性が阻害される場合と、逆に核内に保持されて転写活性が亢進される場合の2種類があります。
インスリン/インスリン様成長因子-1(IGF-1)はPI3K/Aktシグナル伝達系を亢進し、活性化されたAktは転写因子FoxO3aをリン酸化します。この場合、リン酸化されたFoxO3aは核外(細胞質)へ移行して分解されるので、FoxO3aの転写活性は抑制されます。(下図)

図:インスリン/インスリン様成長因子-1(IGF-1)はPI3K/Aktシグナル伝達系を亢進し、活性化されたAktは転写因子FoxO3aをリン酸化する。リン酸化されたFoxO3aは14-3-3というたんぱく質と結合し核外(細胞質)へ移行して分解されるので、FoxO3aの転写活性は抑制される。FoxO3aの標的遺伝子は細胞増殖を停止させ、がん抑制的に作用するので、FoxO3aの核外への移行(不活性化)はがん細胞の増殖を促進することになる。
FoxO3aは細胞周期の進行を阻害するタンパク質p27Kip1の発現を促進します。p27Kip1は細胞周期のG0/G1停止を引き起こすサイクリン依存性キナーゼ阻害因子です。
また、FoxO3aはがん細胞のミトコンドリアに作用してアポトーシスを誘導するタンパク質のBimの発現を亢進することが報告されています(326話)。
つまり、FoxO3aの転写活性を高めることはがん細胞をG0/G1期で細胞周期を止め、アポトーシスを誘導することになります(下図)。

図:インスリンやインスリン様成長因子-1やその他の増殖因子や成長因子はPI3K/Aktシグナル伝達系を活性化し、AktはFoxO3aをリン酸化して核外に移行させて転写活性を阻害(不活性化)する。FoxO3aはサイクリン依存性キナーゼの阻害因子であるp27Kip1やアポトーシスを誘導するBimの発現を亢進してがん細胞をG0/G1期で停止させ、アポトーシスを誘導する。したがって、PI3K/Aktシグナル伝達系の活性阻害は、がん細胞の増殖抑制とアポトーシス誘導に作用する。
【IκBキナーゼはNF-κBを活性化する】
炎症性疾患やがんの治療のターゲットの一つにNF-κB(Nuclear Factor-kappa B、核内因子-κB)いう遺伝子の転写を調節するタンパク質複合体があります。
遺伝子の発現(DNAをmRNAに転写すること)を調節するタンパク質を転写因子と言い、この転写因子はDNA上のプロモーターやエンハンサーといった転写を制御する部分に特異的に結合し、DNAの遺伝情報をmRNAに転写する過程を促進、あるいは逆に抑制する働きを持っています。
NF-κBは転写因子の一つですが、炎症反応や免疫応答や細胞増殖に関連する多くの遺伝子の発現を調節しており、NF-κBの活性化は様々な炎症性疾患やがんを増悪させることが明らかになっています。
NF-κBは免疫グロブリンのκ鎖遺伝子のエンハンサー領域に結合するタンパク質として発見され、当初はリンパ球のB細胞に特異的に発現していると考えられていましたが、後にほとんどの細胞に存在することが明らかになりました。
がん細胞では、NF-κBが活性化すると死ににくくなるので抗がん剤に抵抗性になり、増殖や転移が促進されます。さらに、がん細胞や炎症細胞のNF-κB活性が高まると、内皮細胞増殖因子(VEGF)や単球走化因子-1(monocyte chemoattractant factor-1)やインターロイキン-8(IL-8)やシクロオキシゲナーゼ-2(COX-2)など、腫瘍血管の新生に関与する蛋白質の産生が増加します。
炎症細胞やがん細胞に、炎症性サイトカン(IL-1, TNF-αなど)や酸化ストレス(放射線や活性酸素など)が作用すると、細胞内でNF-κBが活性化されます。このNF-κBを活性化するタンパク質がIκBキナーゼ(IKK)です。
NF-κBは細胞質に存在し、IκB(Inhibitor of κB)と呼ばれる制御蛋白質と複合体を形成し、不活性型で細胞質に局在しています。
炎症性刺激や酸化ストレスやプロテインキナーゼCなどによりIκBのセリン基をリン酸化するIκBキナーゼ(IKK)が活性化されてIκBをリン酸化し、さらに蛋白分解の目印となるユビキチンが結合し、プロテアソームで分解されます。
IκB が外れるとIκBでマスクされていた核内移行シグナルが露出して、NF-κBは核に移行できるようになります。NF-κBはDNA上のκBモチーフ (GGGACTTTCC) と呼ばれる配列に結合し、目的遺伝子の転写活性化を行います。したがって、NF-κBの活性化を阻止することは、炎症性疾患やがんの治療に有効と考えられています。(下図)

図:NF-κB(Nuclear FactorκB)は細胞質に存在し、IκB(Inhibitor of κB)と呼ばれる制御蛋白質と複合体を形成している(①)。炎症性サイトカイン(IL-1やTNF-αなど)や細菌由来のリポ多糖や酸化ストレス(放射線や活性酸素など)はIκBキナーゼを活性化してIκBをリン酸化する(②)。さらにユビキチンが結合してプロテアソームで分解される(③)。IκBが外れるとNF-κB分子内の核内移行シグナルが露出してNF-κBは核に移行し、目的遺伝子の転写を行う(④)。NF-κBで活性化される遺伝子は炎症やがんの進展と関連するものが多い(⑤)。その結果、がん細胞の増殖は亢進し、アポトーシスが起こりにくくなって抗がん剤耐性となり、腫瘍血管の新生が促進される(⑥)。
【IκBキナーゼはFoxO3を不活性化する】
前述のIκBキナーゼ(IκBをリン酸化してNF-κBを活性化する)がFoxO3aをリン酸化して核外へ移行させて不活性化することが報告されています。以下のような論文があります。
IKK-β mediates chemoresistance by sequestering FOXO3; a critical factor for cell survival and death.(IκBキナーゼ-βはFOXO3を不活性化して抗がん剤耐性を引き起こす:細胞の生存と死を制御する重要因子)Cell Signal. 24(6):1361-8, 2012年
FoxO3は細胞の生存や死に関与する遺伝子の発現の制御に関わっています。この論文では、シスプラチンに抵抗性の乳がん細胞MDA-MB-231細胞に比べてFoxO3活性がはるかに高い乳がん細胞MCF-7では、シスプラチン感受性が高いことを示しています。すなわち、FoxO3活性がシスプラチン感受性に重要な因子であることを示しています。
MCF-7細胞ではシスプラチンはFoxO3に依存的な作用機序でアポトーシスを誘導しました。
一方、シスプラチンに抵抗性のMDA-MB-231細胞では、シスプラチン投与後にIκBキナーゼ-β(IKK-β)がFoxO3の活性を阻害し、シスプラチン抵抗性を促進しました。
IKK-βはFoxO3と直接に相互作用をして(リン酸化して)細胞質に移行させ、核内での局在を阻害しました。
乳がん細胞におけるシスプラチン抵抗性はIKK-βの活性化とそれによるFoxO3の不活性化(細胞質への排除)によって起こっているという結論です。したがって、IKK-β活性の阻害は抗がん剤感受性を高める方法として有効だという話です。
IκBキナーゼ(IKK)はIKKαとIKKβとIKKγと呼ばれる調節サブユニットから構成される複合体を形成しています。様々な炎症性シグナルや増殖シグナルに応答してIKK-αとIKK-βはリン酸されて活性化されます。
IKKαとIKKβはアミノ酸配列が極めて似ているにも関わらず、基質特異性と制御様式が異なるため、その機能は大きく異なります。
IKKβ(およびIKKγ)は、腫瘍壊死因子α(TNFα)やリポ多糖(LPS)によるNF-κB活性化に必須です。対照的に、IKKαはIKKβに誘導されたNF-κB活性化を弱める働きをする可能性が指摘されています。
いずれにしても、IKK複合体のうちIKKβのリン酸化は炎症性シグナルに応答して起こり、NF-κBの活性化やFoxO3の不活性化に関与しています。
NF-κBの活性化もFoxO3の不活性化も、ともに増殖を促進しアポトーシスを起こしにくくする作用があります。したがって、IKKβのリン酸化を阻害することは、がん細胞の増殖抑制とアポトーシス誘導に作用することになります。
Aktを欠損させてAktによるFoxO3aの不活性化が起こらないようにした細胞でも、IKKβによってFoxO3aの不活性化が引き起こされます。
つまり、Akt非依存性のFoxO3aの不活性化の機序が存在するので、Akt活性と抑えると同時にIKKβ活性を阻害することが重要です。

図: NF-κBはIκBと複合体を形成して細胞質に存在している(①)。炎症性サイトカイン(IL-1やTNF-αなど)や細菌由来のリポポリサッカライドや酸化ストレス(放射線や活性酸素など)はIκBキナーゼ(IKK)を活性化してIκBをリン酸化する(②)。リン酸化されたIκBは分解され、フリーになったNF-κBは核内に移行し、目的遺伝子の転写を行う(③)。FoxO3aは核内に存在するが、IKKによってリン酸化されると核の外に移行する(④)。核外に移行するとFoxO3aによって転写される遺伝子の発現は阻害される(⑤)。インスリン/インスリン様成長因子-1(IGF-1)はPI3K/Aktシグナル伝達系を亢進し、活性化されたAktはFoxO3aをリン酸化して核外へ移行させる(⑥)。NF-κBで転写が促進される遺伝子は炎症やがんの進展と関連するものが多い(⑦)。FoxO3aによって転写が促進される遺伝子は細胞周期停止やアポトーシス誘導に関連するものが多い(⑧)。したがって、IκBキナーゼ(IKK)やPI3K/Aktの活性化を抑制することは、がん細胞の増殖を抑制し、アポトーシスを誘導する方向で作用する。
【オーラノフィンはIκBキナーゼを阻害する】
オーラノフィン(Auranofin)は、関節リュウマチの治療に使われる経口金製剤で、炎症反応や免疫異常を抑制する作用があります。
最近、オーラノフィンの抗腫瘍効果が注目されてます。
ミトコンドリアのチオレドキシン還元酵素(419話、424話)やIL-6/STAT3経路の阻害(427話)やヒストンアセチル化亢進作用(429話)なども報告されています。
さらにIκBキナーゼ(IKK)の活性を阻害する作用が多数報告されています。以下の論文はその一つです。
Gold compound auranofin inhibits IkappaB kinase (IKK) by modifying Cys-179 of IKKbeta subunit.(金化合物オーラノフィンはIKKβサブユニットのシステイン-179を修飾することによってIκBキナーゼを阻害する)Exp Mol Med. 30; 35(2): 61-6. 2003年
この論文では、オーラノフィンがIκBキナーゼ(IKK)活性を阻害してNF-κBの活性化を阻害する作用に関して、オーラノフィンがIKKのどこに作用するかを検討しています。
IKKαとIKKβの正常な遺伝子と変異型の遺伝子を導入した細胞を使って、オーラノフィンのIKKに対する作用を検討しています。
IKKβの179番目のシステイン(Cys-179)をアラニンに変換した遺伝子を導入した細胞では、オーラノフィンによるIKKβの阻害が見られなくなりました。
つまり、オーラノフィンはIKKβのCys-179の部分に作用して、炎症性シグナルで誘導されるNF-κBとIKKの活性化を阻害することを示しています。
【オーラノフィンはFoxO3aを活性化する】
IKKは核内に存在する転写因子FoxO3aをリン酸化して核外に移行させることによって、FoxO3aの転写活性を阻害することは前述しました。したがって、IKKを阻害するオーラノフィンはFoxO3aを活性化して抗腫瘍作用を示す可能性があります。以下のような論文があります。
Auranofin displays anticancer activity against ovarian cancer cells through FOXO3 activation independent of p53. (オーラノフィンは卵巣がん細胞においてp53非依存性にFOXO3を活性化することによって抗腫瘍作用を示す)International Journal of Oncology. 2014;45(4):1691-1698.
【要旨】
有機金化合物のオーラノフィンは関節リュウマチの治療薬として広く使用されている。新規のがん治療薬のスクリーニングによって、p53欠損の卵巣がん細胞株SKOV3細胞に対してオーラノフィンが強力な抗腫瘍活性を持つことを偶然に発見した。しかしながら、卵巣がんに対するオーラノフィンの抗腫瘍作用の分子メカニズムについてはほとんど解明されていない。
本研究では、オーラノフィンが用量依存的かつ時間依存的にSKOV3細胞の増殖と生存を阻害することを示した。
オーラノフィンはSKOV3細胞において、アポトーシスを引き起こすカスパーゼ-3を活性化し、アポトーシス誘導性タンパク質のBaxとBimの量を増やし、アポトーシス抵抗性にするBcl-2タンパク質の量を減少させた。
さらに、オーラノフィンはIκBキナーゼ(IKK)-βの発現量を低下させ、FOXO3の核内局在を促進し、FOXO3のがん抑制作用を増強し、その結果、SKOV3のアポトーシスを誘導した。
一方、FOXO3の活性を阻害するとSKOV3細胞におけるオーラノフィンのアポトーシス誘導作用は阻止された。
これらの結果は、オーラノフィンはFOXO3の活性化することによって、カスパーゼ-3を介するアポトーシスを引き起こすことを示している。
このp53を欠損した卵巣がん細胞でオーラノフィンがアポトーシス誘導性の遺伝子の発現を誘導しアポトーシスを実行させたことは、オーラノフィンは卵巣がんにおいてp53非依存性のメカニズムでアポトーシスを誘導できることを示している。
卵巣がんは抗がん剤治療が効きやすいのですが、再発すると抗がん剤抵抗性が問題になります。特にがん抑制遺伝子のp53に変異があると、いろんな治療に抵抗性になります。
この論文では、オーラノフィンがp53とは関係ないメカニズムでFoxO3を活性化して抗腫瘍効果を示すことを報告しています。
オーラノフィンで発現が誘導されるBimはアポトーシスを誘導するタンパク質で、FoxO3aによって発現が誘導されることが他の研究でも報告されています。(326話参照)

図:がん細胞において様々な刺激で活性化されたIκBキナーゼβ(IKKβ)はFoxO3aの核内移行を阻止することによってFoxO3aの転写活性を阻害する(①)。FoxO3aはアポトーシス誘導性のBcl-2の発現を阻害し(②)、アポトーシスを誘導するタンパク質(Bax, Bim, Caspase-3など)の発現や活性を高めアポトーシスを誘導する(③)。オーラノフィンはIKKβの活性を阻害する作用によってFoxO3aの転写活性を高めて、がん細胞のアポトーシスを誘導する(④)。
【オーラノフィンとビタミンD3とレチノイド】
ビタミンD3とレチノイドはFoxO3aやサイクリン依存性キナーゼ阻害因子のp21Cip1やp27Kip1の発現を亢進します。(371話参照)
がん細胞では、DNAメチル化やヒストン脱アセチル化によるエピジェネティック(epigenetic)な機序によってがん抑制遺伝子などの遺伝子の転写が抑制されています。
オーラノフィンはヒストン・アセチル化を亢進してがん抑制遺伝子の発現を亢進する作用が報告されています(429話参照)。オーラノフィンがヒストンのアセチル化を亢進する作用によってビタミンDとレチノイドの分化誘導作用を増強することが指摘されています。
さらに、オーラノフィンがIKK(IκBキナーゼ)を阻害してFoxO3aの転写活性を亢進するので、この3つの組合せは、FoxO3aと核内ステロイドホルモン受容体を介してがん抑制性に作用すると言えます。
オールトランス・レチノイン酸は皮膚の角化を促進する作用があるので、多く服用すると口唇の荒れなどの副作用がでてきます。ビタミンDやオーラノフィンと併用する場合は、このような副作用が出て来ないレベルの少量で分化誘導作用は期待できます。
ビタミンD3は体内で活性化されて1,25(OH)2ビタミンD3(Calcitriol)となって作用し、この活性型はフィードバックで制御されているので、副作用がでることは極めて少なく、安価なサプリメントとして利用できます。(371話参照)
オーラノフィンは関節リュウマチの治療薬で安価です。主な副作用は下痢や腹痛や口内炎などの消化器症状が1~5%程度、発疹や掻痒などの皮膚症状が2~3%程度、その他1%以下の頻度で蛋白尿、貧血、浮腫、肝障害などが報告されていますが、比較的副作用の少ない安全性の高い薬です。ただし、重篤な副作用が出る場合もあるので、副作用に十分に注意して使用します。(424話、427話参照)
【オーラノフィンの抗がん作用を高める方法】
がん治療では、副作用が少ない方法でがん細胞の増殖を抑えることができれば理想の治療法になります。
抗がん剤以外の治療に使用されている既存の薬の抗腫瘍効果を利用した「薬の転用(Drug repositioning)」の研究で、多くの候補が見つかっています。
オーラノフィンは関節リュウマチの治療に使用される服用量で、チオレドキシン還元酵素やIL-6/STAT3経路の阻害やヒストンアセチル化亢進作用などの様々な抗がん作用が期待できます。
前述のように、オーラノフィンとビタミンD3とレチノイド(ビタミンA誘導体)のオールトランス・レチノイン酸の併用は、NF-κB活性の阻害とFoxO3aの活性亢進の作用によって、がん細胞の増殖抑制とアポトーシス誘導の作用を発揮します。
しかし、がん細胞はいろんなメカニズムで増殖抑制とアポトーシス誘導に抵抗しようとします。したがって、さらに抗腫瘍効果を高めるために、他の方法を追加する必要がある場合もあります。
炎症性サイトカインの産生抑制やIκBキナーゼの活性阻害作用に関してはサリドマイドの効果が報告されています。
以下のような論文があります。
Inhibition of NF-kappa B activity by thalidomide through suppression of IkappaB kinase activity.(I-κBキナーゼ活性の抑制によるサリドマイドのNF-κB活性の阻害作用)J Biol Chem 276(25): 22382-7, 2001年
【要旨】
かつて鎮静作用や吐気止め作用の薬として使用され、胎児の奇形を引き起こすことが問題になったサリドマイドには、抗炎症作用と抗がん作用を有することが明らかになった。
サリドマイドは炎症性サイトカインの産生を抑制することによって抗炎症作用を示し、血管新生を阻害する作用が抗がん作用の機序と考えられている。
転写因子のNF-κBは、腫瘍壊死因子-α(TNF-α)やインターロイキン-8などの炎症性サイトカインの遺伝子発現を制御している。
関節リュウマチなどの動物実験モデルで、NF-κBの活性を阻害すると炎症を抑制できることが示されている。
本研究では、サリドマイドがI-κBキナーゼの活性を阻害することによってNF-κBの活性を阻害できることを明らかにした。
この結果と一致して、サイトカインによるNF-κBの活性化によって誘導される遺伝子(IL-8、TRAF1、c-IAP2)の発現を阻止した。
以上の結果は、IκBキナーゼ活性を抑制してNF-κBの活性を阻害する作用が、サリドマイドの薬効の作用機序になっていることを示している。
NF-κB/IL-6/STAT3経路を阻害するジインドリルメタン、シクロオキシゲナーゼ-2(COX-2)の活性を阻害するセレコキシブ(celecoxib)の併用も有効です。(428話参照)
アルコールを全く飲まなければ、オーラノフィンとの相乗効果が報告されているジスルフィラムの併用が有効です(419話)。
がん幹細胞に過剰に発現しているアルデヒド脱水素酵素1A1の阻害剤のジスルフィラムはオーラノフィンの抗腫瘍効果を高めます。
進行した肺がんの抗がん剤治療にジスルフィラムを併用すると生存期間を延長する効果が報告されています。(543話参照)
抗がん剤を使わなくても、オーラノフィンなどとの併用で抗腫瘍効果を高めることができると考えられます。
また、低用量の抗がん剤を使ったメトロノミック・ケモテラピー(397話参照)とオーラノフィンを併用すると抗がん作用を強めることができます。
低用量のメソトレキセートとシクロフォスファミドを併用したメトロノミック・ケモテラピーの有効性が報告されていますが、この2つも関節リュウマチの治療に使われています。
糖質制限やケトン食もFoxO3aを活性化します。インスリンはAktの活性を高め、脂肪酸のβ酸化の亢進はFoxO3aの活性を高める作用があります。(下図)

図:糖質を摂取するとインスリンが分泌され、インスリン受容体が活性化されるとホスホイノシチド3-キナーゼ(PI3K)/Aktシグナル伝達系が活性化される。Aktは転写因子のFoxO3aの活性を阻害する。糖質摂取およびインスリンは脂肪酸のβ酸化を阻害する。脂肪酸のβ酸化自体がFoxO3aを活性化することが報告されている。FoxO3aの活性化はがん抑制的に作用する。
ケトン体の一種のβヒドロキシ酪酸がヒストン脱アセチル化酵素を阻害し、Foxo3aのプロモーター領域におけるヒストンの高アセチル化を引き起こし、FoxO3a遺伝子の発現を亢進することが報告されています。(詳しくは322話参照)
血中のβヒドロキシ酪酸が高濃度になるケトン食が、寿命延長やがん抑制に有効である機序として、FoxO3aの活性化が重要な役割を果たしている可能性が指摘されています。
脂肪酸のβ酸化が亢進するだけでFoxO3aの活性化が起こることが報告されています。脂肪酸のβ酸化を促進する中鎖脂肪酸と多く使ったケトン食はFoxO3aをさらに活性化する可能性があります。(詳しくは318話参照)
高脂血症治療薬のフェノフィブラートは様々なメカニズムでFoxO3Aの活性化を引き起こします。
フェノフィブラートが膠芽腫(グリオブラストーマ)に対して抗腫瘍効果を示すことが報告されています。
フェノフィブラートはPeroxisome Proliferator-activated Receptor α(PPARα:ペルオキシソーム増殖因子活性化受容体α)に結合してPPARαを活性化します。
PPARαは核内受容体の一種で、ペルオキシソームを増やして脂肪酸のβ酸化を亢進し、グルコースの解糖系を抑制する働きがあるので、がん細胞のワールブルグ効果(酸素がある条件でも、がん細胞はグルコースの嫌気性解糖系でエネルギーを産生していること)を阻害して、がん細胞の増殖を抑える可能性が指摘されています。
フェノフィブラートはPPARα依存性あるいは非依存性の機序で、PI3K/Aktシグナル伝達系の阻害や、転写因子FoxO3Aの活性化を引き起こします。FoxO3Aの活性化はアポトーシスを誘導するBimの発現を亢進し、グリオブラストーマを死滅させるという機序が提唱されています。(詳しくは326話参照)
【FoxO3aの活性化は多くのがんで有効】
以下のような報告があります。
Pharmacological activation of FOXO3 suppresses triple-negative breast cancer in vitro and in vivo (FOXO3の薬理学的活性化は培養細胞および動物実験の両方でトリプルネガティブ乳がんの増殖を抑制する)Oncotarget. 2016 Jul 5; 7(27): 42110–42125.
【要旨】
トリプルネガティブ乳がんは乳がんの中で最も予後が悪い組織型である。有効な治療法が無いことがトリプルネガティブ乳がんの治療を妨げている。
この研究において、米国食品医薬品局が認可している統合失調症治療薬のトリフルオペラジン(trifluoperazine)と狭心症治療薬のベプリジル(bepridil)が、Aktの473番目のセリン残基(S473)のリン酸化を阻害して、トリプルネガティブ乳がんにおけるFOXO3の核内局在と活性化を促進することを明らかにした。
ベプリジルとトリフルオペラジンはトリプルネガティブ乳がんの培養細胞の増殖を抑制し、動物に移植したトリプルネガティブ乳がんの増殖を抑制した。ベプリジルとトリフルオペラジンのよるトリプルネガティブ乳がん細胞の増殖抑制作用はFOXO3遺伝子の発現を阻害すると抑制された。
ベプリジルとトリフルオペラジンはトリプルネガティブ乳がん細胞におけるがん遺伝子のc-MycとKLF5とドーパミン受容体DRD2の発現を減少させた。ベプリジルとトリフルオペラジンによるc-MycとKLF5とDRD2の遺伝子発現の抑制効果はFOXO3遺伝子の発現を阻害すると消失した。
多くのがん細胞種において、がん遺伝子のc-MycとKLF5とドーパミン受容体DRD2はがん幹細胞の性質を持ったがん細胞を増やすことが報告されているので、ベプリジルとトリフルオペラジンがこれらのたんぱく質の発現量を減少させることは、トリプルネガティブ乳がん細胞におけるベプリジルとトリフルオペラジンによるアポトーシス誘導が、FOXO3依存性の機序によることを示唆している。
トリプルネガティブ乳がんというのは、エストロゲン受容体、プロゲステロン授与歌HER2受容体の3つが陰性の乳がんで、全乳がんの15%くらいを占めます。
トリプルネガティブ乳がんはホルモン療法やハーセプチンが効かないので、進行すると通常の抗がん剤しか無いので、治療法が限定され、死亡率が高い乳がんです。
このトリプルネガティブ乳がんの抗がん剤治療にFoxO3aを阻害する治療を併用すると、抗腫瘍効果を高めることができるということです。
以下のような報告があります。
Reprogramming ovarian and breast cancer cells into non-cancerous cells by low-dose metformin or SN-38 through FOXO3 activation(低用量のメトホルミンあるいはSN-35はFOXO3活性化を通して卵巣がん細胞と乳がん細胞を非がん細胞に再プログラムする)Sci Rep. 2014; 4: 5810.
FOXO3を活性化すると卵巣がんや乳がんのがん細胞が非がん細胞に変わるという論文です。FOXO3を活性化する方法として、低用量のメトホルミンとSN-38を紹介しています。
低用量のメトホルミンあるいはSN-38を投与するとFOXOの核内集積が増加し、DNAダメージマーカーが増え、がん幹細胞のマーカー(CD44、Nanog、Oct-4、c-Myc)が減少することを示しています。その結果、これらの薬は3次元細胞培養における細胞塊(スフェロイド)形成を阻害しました。しかし、FOXO3遺伝子の発現を阻害するとこれらの作用は消失した。
以上の結果は、低用量のメトホルミンあるいはSN-38は、FOXO3の活性化を介して、がん細胞の正常を非がん細胞に変換することを示しています。つまり、副作用が少ない方法でがん細胞の増殖を抑えることが可能になるという話です。
FOXO3を活性化することはがん幹細胞の性状や能力を消失させることになります。FOXO3を活性化すると非がん細胞への再プログラム化が可能という報告です。
メトホルミンは糖尿病治療薬で、前述のようにグリオブラストーマなどのがん細胞を用いた研究でFOXO3を活性化することが報告されています。
SN-38は塩酸イリノテカン(CPT11)の活性型です。CPT-11はプロドラッグであり、まず体内で肝臓に存在するカルボキシルエステラーゼによって強力な抗がん作用をもったSN-38に分解され全身に運ばれて抗がん作用を発揮します。トポイソメラーゼ-1阻害活性を示します。
CPT11は培養細胞に添加しても活性を示さないので、SN-38を使っています。
著者らは、以前の論文で、カンプトテシンがFOXO3の活性化を亢進することを報告しています。そこで、この論文では、FOXO3の活性を亢進さる薬としてメトホルミンとSN-38を使っています。これら2つの薬は、乳がん細胞と卵巣がん細胞で低用量でFOXO3を活性化し、がん細胞の特徴を消失させることを報告しています。
低用量というのは、in vitroの実験でmetformin (100 μM) 、 SN-38 (1 nM)です。
in vivoの実験でmetformin [5 mg/kg body weightd per mouse], SN-38 (10 μg/kg BW/mouse), 週3回で投与しています。この論文ではメトホルミンとSN-38は注射で投与しています。
メトホルミンのマウスで1日に5mg/kgの注射投与は、人間では1mg/kg程度ですが、内服の生体内利用率を考えると5mg/kg程度かもしれません。60kgで300mgですので、通常の250mg製剤を1日1〜2錠で良いように思います。
以下のような報告もあります。
NF-κB–driven suppression of FOXO3a contributes to EGFR mutation-independent gefitinib resistance(NF-κB誘導性のFOXO3a活性の抑制はEGFR変異に非依存性のゲフィチニブ耐性に寄与する)Proc Natl Acad Sci U S A. 2016 May 3; 113(18): E2526–E2535.
ゲフィチニブ(Gefitinib:商品名イレッサ)はEGFRチロシンキナーゼ(上皮成長因子受容体チロシンキナーゼ)阻害剤で肺腺がんの治療に使われます。しかし、ゲフィチニブに反応する肺がんは一部であり、ゲフィチニブが有効性を示す場合でも数ヶ月で耐性ができて効かなくなります。
この論文では、FOXO3a発現レベルとEGFRチロシンキナーゼ阻害剤耐性が逆相関することを報告しています。
つまり、FOXO3a の活性化がEGFRチロシンキナーゼ阻害剤の耐性獲得を阻止する新規の治療法になるということです。
【イベルメクチンはPAK1を阻害してFoxO3aの活性化に寄与する】
細胞の増殖は、増殖因子受容体が細胞外ドメインで増殖シグナルを受け取ることから始まります。
細胞内で機能している多数のシグナル伝達経路の中で、がん細胞の増殖と生存で最も重要なのが、PI3K-Akt経路(生存シグナル経路)とERK-MAPK経路(増殖シグナル経路)です。
細胞膜の増殖因子受容体にリガンド(増殖因子)が結合し2量体化すると、PI3Kのリン酸化活性からAktのリン酸化を通して、アポトーシス(細胞死)の誘導を阻害します。(PI3K-Akt経路)
増殖因子による刺激は、低分子量G蛋白質Rasを経由して、Raf→MEK→ERKとリン酸化反応するMAPK経路(MAPKカスケード)によりシグナルが伝達されます。活性化したERKは最終的に核へ移行し、転写因子が活性化され、細胞増殖関連の遺伝子が発現します。(ERK-MAPK 経路)

図:チロシンリン酸化型受容体にリガンド(増殖因子や成長因子)が結合し2量体化すると、受容体がリン酸化されて活性化する(①)。受容体が活性化されるとPI3Kのリン酸化活性からAktがリン酸化されて活性化する(②)。一方、受容体の活性化は、低分子量G蛋白質Rasを経由して、Raf→MEK→ERKとリン酸化反応するMAPK経路によりシグナルが伝達される(③)。PI3K/Akt経路とMAPK経路の活性化は、最終的に核の転写因子のFoxO3aをリン酸化して不活性化する(④)。FoxO3aの標的遺伝子は、がん細胞の増殖や転移を抑制している(⑤)。リン酸化されたFoxO3aは閣外に移行して、転写活性が阻害される(⑥)。
がん細胞の特徴として、細胞増殖の亢進、細胞死に対する抵抗性、周囲組織への浸潤や遠隔臓器への転移が挙げられます。
正常細胞は、増殖や細胞死や移動は厳密に制御されています。一方、がん細胞はその制御が壊れて、無制限に増殖し、正常組織に浸潤し破壊して、増大していきます。細胞骨格はこれらのプロセスに必須の役割を担っています。
つまり、がん細胞の浸潤や転移は細胞の運動性に依存しており、細胞の運動性には細胞骨格の動的変化が必要です。
細胞の運動性を制御する細胞骨格の動的変化の背後にあるシグナル伝達経路にはRas関連の低分子量GTAアーゼ(Ras-related small GTPases)と、p21活性化キナーゼ(p21-activated kinases :PAKs)を含むエフェクタータンパク質が関与しています。
PAKキナーゼは酵母やショウジョウバエにも存在するセリン/スレオニンプロテインキナーゼのファミリーであり、哺乳類では6つのアイソフォーム(PAK1〜6)が発見されています。
それらはすべて低分子量GTPaseの RacおよびCdc42の直接の標的です。
細胞骨格の動的変化の制御における役割に加えて、PAKは細胞の生存、分裂、遺伝子転写などの様々な細胞活動を調節することが明らかになっています。
いくつかの成長因子受容体チロシンキナーゼ(インスリン、EGF、PDGF、VEGF受容体など)およびGタンパク質共役受容体からのシグナルは、PAKの活性化につながります。
これらの経路は、PI-3キナーゼ(PI3K)とグアニンヌクレオチド交換因子(GEF)の連続的な活性化を通じて低分子量GTPase のRacおよびCdc42を活性化し、PAKを活性化します。
がん細胞では、PAKの活性化は変異Rasを介して頻繁に起こっています。
Rasは最も一般的に変異しているがん遺伝子の1つであり、MAPキナーゼ経路とPI3キナーゼを活性化し、PAKキナーゼを活性化します。

図:受容体チロシンキナーゼ(インスリン受容体、EGF受容体、PDGF受容体、VEGF受容体)およびGタンパク質共役型受容体(G protein coupled receptor : GPCR)からのシグナル(①と②)は、RASおよびPI3Kを活性化し(③)、GDP/GTP交換反応を促進するGEF(guanine-nucleotide exchange factors)の作用によって(④)、Rac/Cdc42はGTP結合型になって活性化し(⑤)、Rac/Cdc42のエフェクターであるp21活性化キナーゼ(PAK-1)を活性化する(⑥)。活性化したPAK-1は細胞の生存と増殖を促進し、がん細胞の増殖と転移を亢進する(⑦)。さらに、PAK-1はRASによるMAPキナーゼ(分裂促進因子活性化タンパク質キナーゼ)経路の活性化を増強する(⑧)。PAK-1の活性化は、RacおよびCdc42を介する以外に、GTPaseに依存しないメカニズムでも起こる。
ヒトがんの70%以上において、PAK-1の活性亢進ががん細胞の増殖に関与していると言われています。
PAK1はAktを活性化してmTORC1を活性化します。
PAK1の阻害剤としてイベルメクチンが有名です。
イベルメクチン(ivermectin)はマクロライド類に属する物質で、腸管糞線虫症や糸状虫や疥癬など多くの寄生虫に有効です。
静岡県伊東市内のゴルフ場近くで採取した土壌から大村智博士により発見された新種の放線菌「ストレプトマイセス・アベルメクチニウス(Streptomyces avermitilis)」が生産するアベルメクチンを元に創製されました。大村智博士はこの発見で2015年にノーベル生理学・医学賞を受賞しています。
イベルメクチンのPAK1阻害作用については674話で解説しています。

図:低分子量Gタンパク質のRASは細胞外のさまざまな刺激(チロシンキナーゼ受容体やサイトカイン受容体やカルシウムチャネルなど)を受けて細胞増殖や生存を促進するRAFキナーゼ(Raf-1)やPI-3キナーゼ(PI3K)など多数のシグナル伝達系を活性化する(①)。p21活性化キナーゼ(PAK-1)はRacおよびCdc42のような低分子量GTPaseによって活性化される(②)。PAK-1はRasシグナル伝達系の主要な経路であるRaf-1/MEK1/ERK経路(③)とPI3K/AKT経路(④)を活性化する。さらに、PAK-1はβ-カテニンをリン酸化してWNT/β-カテニン経路を活性化する(⑤)。このように、PAK-1は複数のシグナル伝達系の制御に関与し、がん細胞の増殖と転移を促進する(⑥)。寄生虫治療薬のイベルメクチン(Ivermectin)はPAK-1とWNT/β-カテニン経路を阻害する作用によって、抗腫瘍作用を発揮する(⑦)。
つまり、イベルメクチンによるPAK1阻害を介したRaf-1/MEK1/ERK経路とPI3K/AKT経路の阻害は、FoxO3aの活性化にも寄与できると言えます。
新刊紹介:膠芽腫(グリオブラストーマ)の根治を目指す補完・代替医療

本書はプリント・オン・デマンド(Print on Demand)という出版形式で書店では販売していません。アマゾンからの購入になります。
アマゾンからの購入はこちらへ:
| « 746)発酵小麦... | 748)オールト... » |