がんの予防や治療における漢方治療の存在意義を考察しています。がん治療に役立つ情報も紹介しています。
「漢方がん治療」を考える
カレンダー
| 2025年2月 | ||||||||
| 日 | 月 | 火 | 水 | 木 | 金 | 土 | ||
| 1 | ||||||||
| 2 | 3 | 4 | 5 | 6 | 7 | 8 | ||
| 9 | 10 | 11 | 12 | 13 | 14 | 15 | ||
| 16 | 17 | 18 | 19 | 20 | 21 | 22 | ||
| 23 | 24 | 25 | 26 | 27 | 28 | |||
|
||||||||
goo ブログ
過去の記事
カテゴリ
最新の投稿
最新のコメント
最新のトラックバック
ブックマーク
プロフィール
検索
gooおすすめリンク



| URLをメールで送信する | |
| (for PC & MOBILE) | |
652)免疫原性細胞死の誘導法(その1):抗がん剤+2-デオキシ-D-グルコースは抗腫瘍免疫を誘導する
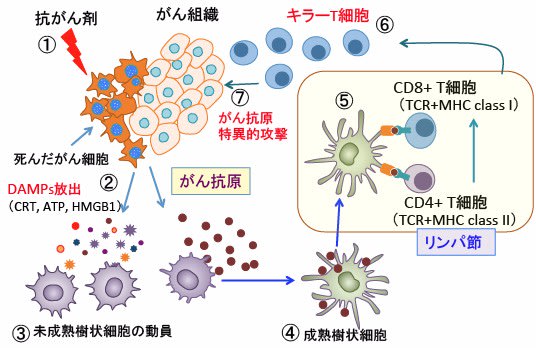
図:抗がん剤(①)によってがん細胞が死滅すると、死滅したがん細胞からカルレチキュリン(CRT)やATPやHMGB1(High-mobility group box 1 protein)などのダメージ関連分子パターン(damage-associated molecular patterns ; DAMPs)が放出される(②)。DAMPsは骨髄や末梢組織から未成熟な樹状細胞をがん組織に動員する(③)。がん組織において死滅したがん細胞から放出されたがん抗原は未熟樹状細胞に取り込まれ、未成熟樹状細胞は活性化されて成熟樹状細胞に分化誘導される(④)。成熟樹状細胞は最寄りのリンパ節に移動し、MHC(Major Histocompatibility Complex)のクラスI及びクラスIIに結合したがん抗原をTCR(T細胞受容体)を介して、CD4+T細胞(ヘルパーT細胞)とCD8+T細胞(キラーT細胞)に提示する(⑤)。がん抗原に反応するキラーT細胞(細胞傷害性T細胞)は抗原提示によって活性化されてクローナルに増殖し(⑥)、がん細胞をがん抗原特異的に攻撃する(⑦)。
652)免疫原性細胞死の誘導法(その1):抗がん剤+2-デオキシ-D-グルコースは抗腫瘍免疫を誘導する
【標準治療の抗がん剤治療は免疫力を犠牲にしている】
標準治療における抗がん剤治療は、最大耐用量(副作用に耐えられる最大量)の抗がん剤を投与することが基本になっています。
患者さんが副作用に耐えられる(死なない)範囲で最大限の投与量を設定するのが最も腫瘍の縮小効果(奏功率)が高いというのが、最大耐用量を投与する根拠になっています。
この方法は白血病や悪性リンパ腫のように抗がん剤が効きやすい腫瘍の場合は有効ですが、抗がん剤が効きにくい腫瘍の場合は、むしろ高用量の抗がん剤投与は、正常細胞のダメージによる副作用が強くなるだけでなく、がん細胞の増殖や浸潤や転移を刺激する可能性が指摘されています。
例えば、高用量の抗がん剤投与によってがん組織が強くダメージを受けると、がん細胞やがん組織の間質にいるがん関連線維芽細胞などからダメージを受けたがん組織を修復するため様々な炎症性サイトカインやケモカインや増殖因子などが産生されます。このような因子によって血管内皮前駆細胞が骨髄から動員されて、血管形成が促進されて、がん細胞の増殖や転移が促進することが明らかになっています。(397話参照)
骨髄の血管内皮前駆細胞はケモカイン受容体のCXCR4を持っているので、がん組織から産生されるケモカインのCXCL12によってがん組織に動員されて血管新生が促進されます(406話参照)。
抗がん剤投与ががん細胞の増殖能や浸潤能を高めることも指摘されています。
さらに、最大耐用量の抗がん剤投与はリンパ球やナチュラルキラー細胞や樹状細胞などの免疫細胞の働きを阻害します。
抗がん剤治療でがん細胞が死滅すると、その死骸を樹状細胞が取り込んで、がん抗原をリンパ球に提示します。抗原特異的な獲得免疫が発動すれば、免疫細胞による腫瘍の排除が促進されます。しかし、高用量の抗がん剤を投与している限り、免疫細胞は働くことができません。(下図)
免疫力を犠牲にして高用量の抗がん剤をやみくもに使用する戦略だけでなく、低用量の抗がん剤と免疫増強を組み合せた戦略も検討する必要があります。

図:高用量の抗がん剤治療によってがん組織がダメージを受けると、ダメージを受けた組織を修復するために、がん組織中にがん関連線維芽細胞が増える(①)。がん関連線維芽細胞からケモカインや増殖因子が産生され(②)、これらの因子は骨髄の血管内皮前駆細胞や炎症細胞をがん組織に動員する(③)。その結果、抗がん剤でダメージを受けたがん組織は血管の新生・増生や炎症性サイトカインの産生、酸化ストレスの亢進が起こる(④)。その結果、がん細胞の増殖が促進され、浸潤や転移が促進される(⑤)。高用量の抗がん剤治療は樹状細胞やリンパ球やナチュラルキラー細胞(NK細胞)などの免疫細胞の働きを阻害する(⑥)。免疫細胞が正常に働かないと、がん細胞の増殖や転移を防ぐことができない(⑦)。
【細胞傷害性T細胞を抑制するPD-1とCTLA-4】
リンパ球の一種のT細胞は、病原菌やがん細胞を攻撃・排除する働きがあります。しかし、T細胞が暴走して正常な細胞を攻撃すると危険なので、いくつかのブレーキ装置が備わっています。これを「免疫チェックポイント」と呼びます。
がん細胞は、ときに巧みにこの免疫チェックポイントを悪用して、T細胞にブレーキをかけてT細胞からの攻撃を逃れようとするのです。がんによるブレーキがかからないようにする薬が免疫チェックポイント阻害薬です。
細胞傷害性T細胞(キラーT細胞)は抗原提示細胞(樹状細胞やマクロファージ)から抗原を提示されると活性化されて、敵(病原菌やがん細胞など)を攻撃します。
細胞傷害性T細胞にはPD-1やCTLA-4という受容体が存在します。PD-1はプログラム細胞死1(programmed death-1)、CTLA-4は細胞傷害性Tリンパ球抗原-4 (cytotoxic T-lymphocyte-associated protein 4)の略です。
これらの受容体のリガンド(受容体に結合して作用する物質)となるPD-L1やB7(B7-1, B7-2)を抗原提示細胞が持つことによって細胞傷害性T細胞の働きを抑制しています。
つまり、PD-1受容体やCTLA-4受容体がリガンドによって刺激されると、T細胞の増殖が停止し細胞死を来すことになります。このようにして細胞傷害性T細胞の過剰な応答を制御しているのです。
細胞傷害性T細胞の働きを阻害するPD-L1やB7はがん細胞にも発現しています。つまり、がん細胞は免疫系の制御システムを利用して、がん組織内の細胞傷害性T細胞の働きを阻止しています。
PD-1受容体やCTLA-4受容体は細胞傷害性T細胞を死滅させるスイッチなようなものなので、これらのスイッチが入らないようにすれば、細胞傷害性T細胞は生き残ってがん細胞の攻撃力を高めることができます。
CTLA-4に対する抗体(ヒト型抗ヒトCTLA-4モノクローナル抗体)のイピリブマブ(ipilimumab: YERVOY)やヒト型抗PD-1モノクローナル抗体のニボルマブ(nivolumab商品名「オプジーボ(Opdivo)」)などがあります。
このような免疫チェックポイント阻害剤を使用すると、がん細胞を攻撃する細胞傷害性T細胞の働きを高めることが可能になります。
体に備わったがん細胞に対する攻撃力を高めてがんを治療しようというのが「がんの免疫療法」の理論です。「免疫細胞を活性化する」という従来の免疫療法では十分な効果が得られなかったのですが、その大きな理由は免疫応答にブレーキをかける仕組みの存在です。このブレーキを解除して免疫細胞に100%の力でがん細胞を攻撃させようというのが、CTLA-4やPD-1/PD-L1をターゲットにした治療法です。(下図)
ただ、この治療法は免疫細胞の暴走を許して、自己免疫疾患を引き起こすという副作用もあります。

図:抗原提示細胞上にはMHCクラスII(MHC-II)といわれる分子があり、抗原を介してT細胞上のTCR(T細胞受容体)と反応して細胞傷害性T細胞を活性化する(①)。T細胞上にはCD28とCTLA-4があり、CD28は恒常的に発現し、抗原提示細胞からのB7-1やB7-2というリガンドによってT細胞活性化に作用する(②)。一方、CTLA-4はT細胞活性化にともなって発現が誘導され、B7-1やB7-2によって刺激されるとT細胞を抑制する(③)。CTLA-4はCD28よりもB7に対する親和性が強いので、活性化したT細胞の過剰な応答を抑制する。同様に、PD-1(Programmed death-1)は抗原提示細胞のPD-L1(別名B7-H1)と結合することによって抑制型の免疫調節シグナルを活性化させる(④)。がん細胞もB7-1やB7-2やPD-L1が発現しており、細胞傷害性T細胞の働きを抑制している。T細胞のCTLA-4とPD-1の働きを特異抗体で阻害すると、がん細胞に対する細胞傷害性T細胞の働きを高めることができる(⑤)。
【免疫チェックポイント阻害薬+化学療法で生存率が向上】
細胞毒性によって効果を発揮する通常の抗がん剤治療は、急性白血病や悪性リンパ腫や精巣腫瘍のように一部の悪性腫瘍を根治することはできますが、肺がんや膵臓がんなど多くの固形がんに対しては、その効果は極めて限定的です。
このような固形がんの治療においては、「最終的にがんを治すのは免疫力」という考え方が重要になってきます。そこで、抗がん剤治療と免疫療法の併用が検討されています。
抗がん剤治療と免疫療法の併用については、異なる考え方が存在します。
一つは、「抗がん剤は免疫細胞の働きを弱めるので、免疫療法と抗がん剤は併用すべきでない」という意見です。
もう一つは、「抗がん剤治療で免疫力が低下するので、その低下を補う方法として免疫療法の併用は有効」という意見です。
どちらの意見も論理的に納得できますが、言っていることは全く真逆です。
最近は第3の考え方が指摘されています。それは「抗がん剤が抗腫瘍免疫を誘導するので、抗がん剤と免疫療法を併用する方が良い」という考え方です。
前述のように、リンパ球などの免疫細胞へのダメージが強い「最大耐用量」の抗がん剤を使う場合は、免疫療法の効き目は期待しにくいように思います。しかし、適度な抗がん剤治療は免疫療法の効き目を高めることが最近の研究で明らかになっています。
オプジーボなどの免疫チェックポイント阻害剤と抗がん剤治療を併用すると、免疫チェックポイント阻害剤単独よりも生存期間を延ばすことが明らかになっています。
免疫チェックポイント阻害薬である抗PD-1抗体ペムブロリズマブ(商品名キイトルーダ)は2016年12月に「PD-L1陽性の切除不能な進行・再発の非小細胞肺がん」への使用が承認されています。つまり、免疫チェックポイント阻害薬の単剤での有効性が示されています。
さらに、2018年4月には、未治療の非扁平上皮の非小細胞肺がんに対して、抗がん剤治療+ペムブロリズマブ併用療法の有効性が報告されています。
これは、KEYNOTE-189という第Ⅲ相二重盲検ランダム化比較試験の結果で明らかになっています。
KEYNOTE-189試験では、2016年2月~17年3月に日本を含む16カ国118施設で登録された616例を化学療法+ペムブロリズマブ群(410例)と化学療法+プラセボ群(206例)に2:1でランダムに割り付け、化学療法はペメトレキセド(アリムタ)+プラチナ製剤が使用されています。
中央値で10.5カ月の追跡の結果、12カ月時点の全生存率はプラセボ群の49.4%(95%CI 42.1~56.2%)に対し、ペムブロリズマブ併用群では69.2%(同64.1~73.8%)でした。全生存期間の中央値は、プラセボ群の11.3カ月(95%CI 8.7~15.1カ月)に対し、ペムブロリズマブ併用群では未到達であり、死亡リスクが有意に51%減少していました〔死亡のハザード比(HR)0.49、95%CI 0.38~0.64、P<0.001〕。
無増悪生存期間の中央値はプラセボ群の4.9カ月(95%CI 4.7~5.5カ月)に対し、ペムブロリズマブ併用群では8.8カ月(同7.6~9.2カ月)と約2倍の有意な延長が認められました。
これらの結果から、「未治療の転移性非扁平上皮NSCLCでEGFR 変異またはALK 変異が陰性の患者に対して、ペメトレキセドおよびプラチナ製剤による標準化学療法にペムブロリズマブを併用することにより、化学療法単独に比べて全生存期間、無増悪生存期間の有意な延長が示された」と結論しています。
腫瘍におけるPD-L1の発現割合にかかわらず、ペムブロリズマブ併用群が化学療法単独群に対して有意に全生存期間の延長を示しています。
ペムブロリズマブ併用群では、プラチナ製剤とペメトレキセドを3週間隔で4回投与後も、ペメトレキセドについては原則的に継続して併用されています。
免疫チェックポイント阻害薬はTリンパ球を中心とする免疫系の細胞を活性化してがんを攻撃する治療薬です。一方、化学療法は骨髄抑制を来し、免疫系細胞を疲弊させると考えられています。
しかし、本試験における化学療法は3週間隔で行われるため、一過性に骨髄抑制を生じても、次回投与の直前には骨髄抑制からリンパ球をはじめとする免疫系の細胞は回復してきます。
化学療法と免疫チェックポイント阻害薬の併用効果については、異なる作用機序の薬剤が単に細胞傷害において相加的に働いているだけでなく、免疫学的に「相乗的」に働いている可能性が示唆されています。
つまり、抗がん剤治療ががん細胞に対する免疫応答を高める可能性が指摘されています。
【抗がん剤はがん細胞の免疫原性細胞死を誘導する】
抗がん剤はがん細胞を死滅して、抗腫瘍免疫を誘導する作用があることが指摘されています。免疫療法を行うときに抗がん剤をうまく利用すると、抗腫瘍免疫を増強できるという考えです。 以下のような論文があります。
Immunogenic effects of chemotherapy-induced tumor cell death.(化学療法による腫瘍細胞死の免疫原性効果)Genes Dis. 2018 May 17;5(3):194-203.
【要旨】
従来の化学療法の臨床的効果が腫瘍細胞への毒性のみに起因するのではなく、免疫監視機構の活性化にも起因することが、最近の多くの動物実験や臨床研究によって示唆されている。抗がん剤による免疫監視機構の活性化というメカニズムは過去の研究においてほとんど無視されてきた。
抗腫瘍免疫応答は、免疫原性細胞死(immunogenic cell death)が引き金になって誘導される。この免疫原性細胞死は、カルレチキュリン(calreticulin)の細胞表面移行、ATPおよびhigh mobility group box 1 (HMGB1)タンパク質の細胞外放出、タイプ1のインターフェロン応答の刺激によって特徴付けられる細胞死である。
ここでは、従来の化学療法剤が免疫原性細胞死誘導剤として作用し、免疫抑制性の微小環境内で腫瘍浸潤リンパ球の働きを調節し、抗腫瘍免疫を再活性化できることを示す最近の研究を要約する。
通常の化学療法のこのような免疫学的効果は、がん患者の予後をより良くする可能性がある。さらに、免疫原性細胞死を誘発する化学療法と免疫療法との組み合わせは、がん患者の臨床転帰を改善するための有望なアプローチである。
がんは無限に増殖する細胞の病気と考えられてきました。したがって、がん細胞の増殖のメカニズムを解明し、増殖を阻害することが抗がん剤治療の目標となってきました。
しかし、生体には免疫監視機構があり、免疫システムががん細胞を排除しています。しかしながら、がん細胞は免疫監視機構から逃避するメカニズムを使って、排除を回避しています。
がん細胞の増殖は単にがん遺伝子の活性化やがん抑制遺伝子の不活性化だけで起こるものではありません。
がん組織の微小環境ががん細胞の増殖に影響しています。特に、免疫細胞のはたらきが重要です。
免疫細胞にはがん細胞を攻撃するキラーT細胞やNK細胞だけでなく、免疫システムを抑制する制御性T細胞、骨髄由来抑制細胞などがあります。
免疫を抑制するメカニズムは、T細胞の暴走による正常細胞での攻撃を避けるために存在します。しかし、がん細胞はこのような免疫抑制性のメカニズムを利用して、T細胞からの攻撃を回避しています。
免疫チェックポイント阻害剤などで、がん細胞の免疫監視機構回避のメカニズムを阻止できれば、がん細胞を免疫の力で排除できます。その結果、転移がんでもがんを根治できる可能性が出てきます。
【ダメージ関連分子パターンが免疫応答を刺激する】
体を構成する正常細胞は毎日多数の細胞がアポトーシスで死滅し、組織幹細胞が細胞分裂して組織の細胞を供給しています。
このような生理的な死に対して、体がいちいち反応して炎症や免疫応答を行えば、大変なことになります。しかし、このような生理的な細胞死は、炎症や免疫応答を引き起こさない死に方をするので、問題は起こりません。
一方、何らかのダメージやストレスで細胞が傷害されたときは、それを認識して対応する必要があります。
例えば、神経が熱や痛みを感じるようになっているのは、体に危害を与える傷害を認識してそれを避ける必要があるからです。
同様に、細胞がダメージを受けたとき、そのような細胞からは通常であれば細胞内に隠れている成分が放出され、炎症細胞や免疫細胞を活性化するメカニズムが存在します。
このような炎症を引き起こす細胞内にある成分をDAMPs(damage-associated molecular patterns; ダメージ関連分子パターン)と総称しています。
細胞傷害に伴って細胞から放出され、周囲の組織や細胞に危険を知らせるアラームのような役割を担う因子のことです。
DAMPsが細胞外や細胞膜上に露出するような細胞死が起こると、炎症細胞(マクロファージや好中球など)やリンパ球や線維芽細胞などが動員され、炎症反応が引き起こされ、ダメージを受けた組織の修復が起こります。
このメカニズムは自己免疫疾患などの慢性炎症性疾患の原因ともなります。
しかし、抗がんや放射線を使ったがん治療の場合は、このダメージ関連分子パターン(DAMPs)を誘導する細胞死のメカニズムを利用すると、がん特異免疫を増強できることが知られています。
DAMPsは骨髄や末梢組織から未成熟な樹状細胞をがん組織に動員し、樹状細胞は死滅したがん細胞から放出されたがん抗原によって活性化され成熟します(トップの図)。
つまり、放射線照射や一部の抗がん剤が免疫原性の高い細胞死を誘導することが知られており、このような細胞死をもっと効率的に行う手段があれば、がん治療の効果を高めることができます。
DAMPsは、細胞質や核やミトコンドリアや小胞体などに存在する成分が放出されたもので、炎症細胞や免疫細胞を刺激します。
DAMPsとしては、ミトコンドリアのATP, DNA, フォルミルペプチド、核のヒストンやHigh-mobility group box 1 protein(HMGB1)、High-mobility group nucleosome binding protein 1(HMGN1)、細胞質のATPやF-アクチン、小胞体のカルレチキュリン(Calreticulin)などが知られています。(下図)
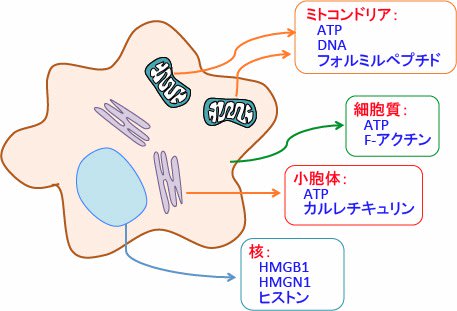
図:細胞がダメージを受けたとき、通常であれば細胞内に隠れていた成分が放出され、炎症細胞や免疫細胞を活性化するメカニズムが存在する。このような炎症を引き起こす細胞内にある成分をDAMPs(damage-associated molecular patterns; ダメージ関連分子パターン)と総称している。DAMPsは、細胞質や核やミトコンドリアや小胞体などに存在する成分が放出されたもので、炎症細胞や免疫細胞を刺激する。DAMPsとして、ミトコンドリアのATP, DNA, フォルミルペプチド、核のヒストンやHigh-mobility group box 1 protein(HMGB1)、High-mobility group nucleosome binding protein 1(HMGN1)、細胞質のATPやF-アクチン、小胞体のカルレチキュリン(Calreticulin)などが知られている。
抗がん剤治療が免疫原性細胞死を誘導することが知られています。その作用は、抗がん剤の種類や投与量によって異なるようです。
免疫原性細胞死を誘導できる抗がん剤として多くのアントラサイクリン系薬(doxorubicin, epirubicin, idarubicinなど)、mitoxantrone、 オキサリプラチン(oxaliplatin)、シクロフォスファミド(cyclophosphamide) 、bortezomibなどが報告されています。
例えば、大腸がんや乳がんでは、アントラサイクリンやオキサリプラチンががん組織内のキラーT細胞の数を増やし、免疫抑制性に作用する制御性T細胞の数を減らすことが報告されています。
従来、抗がん剤の抗腫瘍効果は、がん細胞を直接死滅させる機序によると考えられてきました。しかし、免疫応答を誘導して、免疫監視機構を活性化する機序も関与していることが指摘されるようになったのです。
したがって、抗がん剤治療と免疫療法を積極的に併用する治療も根拠があります。
漢方治療が抗がん剤治療の成績を高める理由も、免疫力の増強が関与していると考えられます。
【小胞体ストレスを引き起こす薬物+抗がん剤=免疫原性細胞死】
抗がんや放射線を使ったがん治療の場合は、ダメージ関連分子パターン(DAMPs)を誘導する細胞死のメカニズムを利用すると、がん特異免疫を増強できることが知られています。
すなわち、DAMPsを誘導しやすくする薬剤は抗がん剤や放射線治療の抗腫瘍効果を高めることができます。
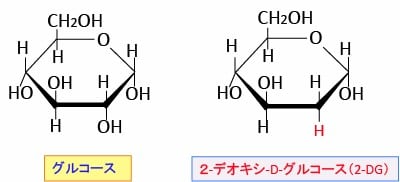
2-デオキシ-D-グルコース(2-Deoxy-D-Glucose:2-DG)は小胞体ストレスを高めてがん細胞を死滅させる作用も報告されています。さらに、2−デオキシ-D-グルコースは抗がん剤や放射線の免疫原性細胞死を増強することが報告されています。
2-デオキシ-D-グルコース(2-Deoxy-D-Glucose:2-DG)はグルコース(ブドウ糖)の2位のOHがHに変わっているグルコース類縁物質です。
小胞体(Endoplasmic reticulum)は、細胞内における分泌・膜タンパク質の品質管理において大切な小器官です。
2-DGは解糖系を阻害する以外に、タンパク質に糖鎖が着くN-グリコシル化の過程を阻害するので、糖タンパク質の生成を阻害します。
グリコシル化というのはタンパク質に糖類が付加する反応で、小胞体で行われて、正常に糖が付加したタンパク質はゴルジ体に運ばれます。
糖鎖異常の糖タンパク質は、折り畳みが不完全な異常タンパク質になり、小胞体に蓄積して小胞体ストレスを引き起こし、細胞死の原因にもなります。
2-デオキシ-D-グルコース(2-DG)は小胞体ストレスを高めて免疫原性細胞死を増強する作用が知られています。
2-DGはグルコースと同じトランスポーター(輸送担体)で取り込まれるので、細胞内の取込みの段階でグルコースの拮抗阻害剤として作用します。
細胞内では、ヘキソキナーゼによってリン酸化されて、2-デオキシグルコース-6リン酸(2-DG-6リン酸)に変換されますが、この2-DG-6リン酸は解糖系の先の代謝系には進めない(ヘキソキナーゼの先の解糖系酵素で代謝できない)ので、ATP産生量が減ります。さらに、蓄積した2-DG-6リン酸はヘキソキナーゼを阻害する作用もあるので、正常なグルコースの代謝も阻害されます。
さらに、2-DGはタンパク質のN-グリコシル化(N-glycosylation)を阻害するので小胞体ストレス応答を誘導します。
2-DGとエトポシドの併用で、免疫原性細胞死を誘導し、樹状細胞が活性化され、CD8+の細胞障害性T細胞の活性が亢進することが報告されています。
抗がん剤でがん細胞を死滅させるときに2−DGを投与しておくと、死滅したがん細胞は免疫原性が高くなるので、がん抗原特異的な抗腫瘍免疫を誘導でき、延命効果を高めることができるというメカニズムです。
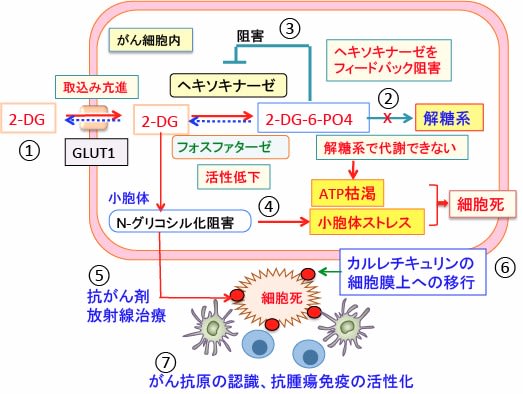
図:2-デオキシ-D-グルコース(2-DG)はグルコース(ブドウ糖)の2位のOHがHに変わっているグルコース類縁物質で、グルコースと同様にグルコーストランスポーター(GLUT1)によって細胞内に取り込まれる(①)。細胞内のヘキソキナーゼで2-DG-6リン酸になるが、それから先の解糖系には進めない(②)。さらに2-DG-6リン酸はヘキソキナーゼを阻害するので、解糖系でのATP産生を阻害する(③)。2-DGは小胞体でのタンパク質のN-グリコシル化(糖鎖の結合による修飾)を阻害し、小胞体ストレスを引き起こす(④)。この状態で、抗がん剤や放射線でがん細胞にダメージを与えると(⑤)、小胞体のカルレチキュリンというタンパク質が死滅したがん細胞の細胞膜上に移行し(⑥)、「ダメージ関連分子パターン(danger-associated molecular patterns:DAMP)」として免疫細胞に認識され、抗腫瘍免疫が活性化される(⑦)。
2-DGと抗がん剤(シクロフォスファミドやエトポシドやアントラサイクリン系)の併用投与による抗腫瘍免疫応答を活性化する治療と、抗原提示細胞やCTLの活性を増強する治療を交互に、あるいは併用して行えば、免疫系による腫瘍細胞の排除ができる可能性が高くなります。
2-DGと抗がん剤を併用すると、小胞体のカルレチキュリン(Calreticulin)というタンパク質が細胞膜上に露出して免疫原性を高めるという結果が報告されています。
以下のような報告があります。
Combination of glycolysis inhibition with chemotherapy results in an antitumor immune response.(抗がん剤治療に解糖系阻害を併用すると抗腫瘍免疫応答が引き起こされる)PNAS 109 (49): 20071-20076, 2012年
【要旨】
細胞のDNAにダメージを与える抗がん剤の多くは、抗腫瘍免疫を誘導する作用がある。解糖系の亢進はがん細胞の最も良く知られた特徴の一つである。そこで、解糖系を阻害する2-デオキシグルコース(2-DG)と細胞傷害性の抗がん剤を併用した場合、抗腫瘍免疫の誘導にどのような影響を及ぼすかを検討した。
2-DGと抗がん剤のエトポシドは、免疫機能の正常なマウスにおいては相乗的に作用して寿命を延長した。しかし、免疫機能不全のマウスに対しては寿命延長効果は認められなかった。
2-DGとエトポシドの両方を投与したマウスにおいてのみ、がん細胞特異的なT細胞の十分な活性化が認められた。
さらに、2-DGとエトポシドの両方の処理によって死滅したがん細胞をマウスに免疫すると、同じ腫瘍の再度の移植に対して拒絶した。
この効果の少なくとも一部は、細胞膜上のERp57/calreticulinの出現が関連していた。
これらの結果は、がん細胞の解糖系をターゲットにすると、死滅がん細胞による通常の免疫寛容誘発性の刺激を、腫瘍免疫誘発性の刺激に変換できることを示している。このメカニズムを利用すると免疫化学療法の新しい戦略を作りだすことができる。
小胞体(Endoplasmic reticulum)は、細胞内における分泌・膜タンパク質の品質管理において大切な小器官です。
カルレチキュリンは、小胞体内腔における主要なカルシウム結合(蓄積)タンパク質として機能する多機能タンパク質です。分子シャペロンとして分泌タンパク質の品質管理の働きも行っています。また核では転写調節の働きを行っています。
2-DGは解糖系を阻害する以外に、タンパク質に糖鎖が着くN-グリコシル化の過程を阻害するので、糖タンパク質の生成を阻害します。
グリコシル化というのはタンパク質に糖類が付加する反応で、小胞体で行われて、正常に糖が付加したタンパク質はゴルジ体に運ばれます。
糖鎖異常の糖タンパク質は小胞体に蓄積して小胞体ストレスを引き起こし、細胞死の原因にもなります。(小胞体ストレスについては298話参照)
2-DGの場合は、糖タンパク質のグリコシル化が阻害され、小胞体ストレスが起こり、その状態で死滅すると死滅した細胞の細胞膜の表面にカルチキュリンが移行してダメージ関連分子パターンとなり、免疫細胞を活性化する結果、抗腫瘍免疫が活性化されるということです。

図:細胞がダメージを受けて死滅するとき、細胞内に存在する成分が放出されて炎症細胞や免疫細胞を刺激する。ミトコンドリアのATPや核のHMGB1(High-mobility group box 1 protein)は細胞外に放出されると樹状細胞を刺激する(①)。小胞体のカルレチキュリン(Calreticulin)は細胞表面に出て、樹状細胞に認識され、貪食のシグナルとなり、がん抗原を提示する働きを活性化する(②)。 2-DG(2-デオキシ-D-グルコース)は、タンパク質に糖鎖が着くN-グリコシル化の過程を阻害するので、糖鎖異常の糖タンパク質が小胞体に蓄積して小胞体ストレスを引き起こす(③)。小胞体ストレスの高い状態で放射線や抗がん剤でがん細胞が死滅するとカルレチキュリンが多く露出した死細胞となる(④)。このような免疫応答を引き起こしやすい細胞死を「免疫原性細胞死」という(⑤)。放射線治療や抗がん剤治療に2-DGを使用すると免疫原性細胞死を誘導してがん抗原に特異的な抗腫瘍免疫を高めることができる。
【低用量のシクロフォスファスファミド単独で大きな腫瘍が消滅する】
シクロフォスファミドのメトロノミック投与が、抗腫瘍免疫を活性化して、大きな腫瘍を消滅できるという実験結果も報告されています。以下のような報告があります。
Metronomic cyclophosphamide eradicates large implanted GL261 gliomas by activating antitumor Cd8+ T-cell responses and immune memory.(シクロフォスファミドのメトロノミック投与は、抗腫瘍性のCD8陽性T細胞応答と免疫記憶を活性化することによって、大きなGL261グリオーマ移植腫瘍を消滅させる)Oncoimmunology. 2015 Feb 18;4(4):e1005521. eCollection 2015.
【要旨】
細胞毒性のある抗がん剤治療は免疫原性細胞死を誘導する。しかし、大きな腫瘍を免疫細胞だけの作用で縮小させ、しかも長期間の免疫記憶を成立させるために有効な方法は確立されていない。
本研究では、この問題を検討するために、免疫系が正常なマウスにGL261グリオーマ細胞を移植した実験系を用い、6日置きのシクロフォスファミドのメトロノミック投与の効果を検討した。
シクロフォスファミドの6日おきのメトロノミック投与の2サイクルの治療で、腫瘍細胞特異的なCD8陽性細胞傷害性T細胞(CTL)とナチュラルキラー(NK)細胞、マクロファージ、その他の免疫細胞を増やし、活性化した。
このようなCTLやNK細胞といったエフェクター細胞はシクロフォスファミド投与の6日後にピークになりその後減少した。制御性T細胞の数はCTLやNK細胞と逆の動きを示した。
間歇的なシクロフォスファミドを数回繰り返すことによって腫瘍は縮小し、消滅した。
腫瘍の消滅にはCD8陽性キラーT細胞(CTL)が必要であった。GL261細胞の再移植に対して、末梢血中のCTLの増加と腫瘍組織でのCTLの浸潤が認められ、抗原特異的な免疫記憶が成立していた。
以上の結果から、抗がん剤のシクロフォスファミドの単独の投与でも、その投与量と投与スケジュールを適切化すれば、大きな腫瘍を縮小させ、さらに消滅させ、免疫記憶を成立させることも可能であることが示された。
シクロフォスファミドを使ったメトロノミック・ケモテラピーは血管新生阻害作用によって抗腫瘍効果を示すと考えられています。しかしながら、最近の研究では、自然免疫の活性化など免疫機能を介したメカニズムの関与が指摘されています。
この研究グループは、がん抗原特異的なキラーT細胞の活性化と移植腫瘍の縮小に、シクロフォスファミドの間歇的なメトロノミックな投薬スケジュールが有効だと報告しています。

図:メトロノミック・ケモテラピー(Metronomic Chemotherapy)は低用量の抗がん剤を頻回に投与する(①)。低用量の抗がん剤は腫瘍免疫を抑制する制御性T細胞(Treg)や骨髄由来抑制細胞(MDSC)の活性を阻害する作用がある(②)。その結果、がん細胞を排除するキラーT細胞など抗腫瘍免疫が増強される(③)。低用量の抗がん剤は血管新生を阻害する作用がある(④)。このように、規則的に低用量の抗がん剤を頻回に投与していくメトロノミック・ケモテラピーに、自然免疫や獲得免疫を活性化する方法を併用すると、免疫機序でがんを排除できる可能性が高まる。
シクロフォスファミドで死滅すると免疫細胞が認識しやすい免疫原性細胞死を誘導します。
高用量だと、免疫原性細胞死を誘導しますが、免疫系は抑制されます。また、がん組織がダメージを受けると血管新生が促進され、がん組織の増大を招く場合もあります。
一方、低用量のメトロノミック投与の場合、免疫原性細胞死は起こりませんが、血管新生が起こらず、骨髄由来抑制細胞(MDSC)と制御性T細胞(Treg)の活性は抑制され、細胞傷害性T細胞(CTL)やナチュラルキラー(NK)細胞は抑制されないので、抗原特異的な抗腫瘍免疫を活性化することができます。
つまり、低用量のメトロノミック投与は、最大耐用量を投与する通常の抗がん剤治療とは異なるメカニズムで腫瘍縮小効果を発揮します。
しかも、免疫記憶が成立するので、再発を予防できることになります。

図:抗がん剤のシクロフォスファミドは高用量でがん細胞の免疫原性細胞死を引き起こすが、低用量では細胞死は誘導できない(①)。腫瘍組織の血管新生は低用量で阻害されるが、高用量では、細胞死によって産生されるサイトカインや増殖因子によってむしろ血管新生が促進される(②)。T細胞の働きを抑制する骨髄由来抑制細胞(MDSC)と制御性T細胞(Treg)はシクロフォスファミドの低用量で活性が抑制され、高用量でも細胞死によって活性が抑制される(③)。低用量ではNDSCとTregの活性低下によって、細胞傷害性T細胞(CTL)とナチュラルキラー細胞(NK細胞)の活性は亢進するが、高用量では細胞死によって抑制される(④)。これらの総合作用によってシクロホスファミドは、低用量ではがん細胞の増殖を抑制し(⑤)、高用量では増殖を促進する(⑥)。
【短期間歇的な抗がん剤投与による免疫原性細胞死の誘導】
通常の抗がん剤治療はがん細胞を死滅させることを第一の目標とし、がん組織の縮小(奏功率)を指標にして効果を評価しています。
その結果、殺細胞作用の強い抗がん剤や複数の抗がん剤の組合せが行われるようになり、がん細胞の増殖を抑えるために、抗がん剤治療でがん細胞を攻撃し続けるという戦略を取ります。その結果、がんが存在する限り、抗がん剤治療を行うことになります。
しかし、作用の強い抗がん剤治療で一時的に腫瘍が縮小しても、その効果は長続きせず、いずれがん細胞が耐性を獲得したり、患者側の体力低下などで、抗がん剤治療自体が行えない状況になります。
放射線照射やある種の抗がん剤は、抗腫瘍免疫を刺激できる細胞死(免疫原性の高い細胞死)を引き起こします。しかし、免疫原性細胞死を誘導しても、最大耐用量の抗がん剤を使用し続ければ、リンパ球や樹状細胞も増殖を阻害されるので、抗腫瘍免疫は発動も活性化もできません。
そこで、免疫原性細胞死を引き起こす抗がん剤投与を短期間で間歇的に行いながら、抗がん剤の休薬期間に樹状細胞やリンパ球(ヘルパーT細胞やキラーT細胞)を活性化し、免疫抑制性の因子(骨髄由来抑制細胞、制御性T細胞、プロスタグランジンE2など)を抑制する治療を行って積極的に抗腫瘍免疫を活性化すれば、がん組織の消滅も不可能ではありません。
通常の抗がん剤治療は体の治癒力や免疫力を犠牲にしている点が最も問題だと思います。免疫細胞ががん細胞を攻撃しやすい条件と微小環境を作るという治療法も検討する価値があります。
つまり、放射線照射や短期間の抗がん剤治療によってがん細胞の免疫原性細胞死を誘導し、その後に抗腫瘍免疫を高める治療法を行うという治療法です。
このような短期間の間歇的な抗がん剤治療としては、昔から使用されて比較的安価なシクロフォスファミドとエトポシドが有用です。両方とも経口投与で、免疫原性細胞死を誘導することが知られています。
【シクロフォスファミドとエトポシド】
強い抗がん剤治療で効果が無くなった患者さんを対象に、昔から使用されている安価な抗がん剤(シクロフォスファミド、エトポシドなど)の治療効果が最近多く報告されています。シクロフォスファミドとエトポシドの使われ方を知る上で参考になります。
Oral etoposide monotherapy is effective for metastatic breast cancer with heavy prior therapy.(経口のエトポシドの単剤治療は高度な抗がん剤治療を受けた転移性乳がんに有効)Chin Med J (Engl). 2012 Mar;125(5):775-9.
【要旨】
研究の背景:頻回の抗がん剤治療をすでに受けている転移性乳がん患者の治療選択は限られている。このような患者に経口エトポシドが効果を示すことが報告されている。しかしながら、高度の抗がん剤治療を受けているがん患者は、体力や抵抗力の低下も強く、長期間の経口エトポシド投与には限界もある。この研究では、すでに高度の抗がん剤治療を受け、効果を認めなくなった転移性乳がんの中国人患者を対象に、短期間の経口エトポシド投与の有効性と安全性を評価する臨床試験を行った。
方法:少なくとも2種類の投与法での抗がん剤治療を受けてた転移性乳がん患者を対象に、エトポシド(60mg/m2/day)を1〜10日まで投与し、その後11日間は休薬するスケジュールで治療を行った。無増悪生存期間、奏功率、臨床的有用性、全生存期間、副作用を評価した。
結果:32人の患者が経口エトポシドの投与を受け、投与サイクルの中央値は6サイクル(2〜20サイクル)であった。8例(25%)は部分奏功、14例が病状安定であった。他覚的奏功率(objective response rate)は25%であった。9例が24週間以上の病状安定(SD)で臨床的有効性(clinical benefit rate)は53%であった。
無増悪生存期間の中央値は5ヶ月(1.5〜17ヶ月)、全生存期間の中央値は16ヶ月(3〜51ヶ月)であった。
臨床的有用性を認めた患者では、そうでない患者に比べて生存期間の延長を認めた(25ヶ月vs 11ヶ月)
この治療の前に4種類以上の投与法による抗がん剤治療を受けていた16例においては、4例が部分奏功、4例が24週間以上の病状安定を認め、臨床的有用性は50%であった。
血液学的な副作用として貧血(43.8%)、白血球減少(38.5%)であった。吐き気/嘔吐(75.0%)、脱毛(62.5%)が最も多い副作用であった。
結論:高度に抗がん剤治療を受けた転移性乳がんの患者に経口エトポシド治療は有効であり、副作用も耐えられるレベルであった。
同じ著者から同じプロトコールでの多施設第2相試験の結果が報告されています。
Efficacy of oral Etoposide in pretreated metastatic breast cancer: a multicenter phase 2 study.(治療を受けた転移性乳がんにおける経口エトポシドの有効性:多施設第2相試験)Medicine (Baltimore). 2015 May;94(17):e774.
【要旨】
アントラサイクリン系とタキサン系の抗がん剤治療に抵抗性になった転移性乳がん患者に有効な抗がん剤治療法はない。このような治療抵抗性の転移性乳がん患者における経口エトポシド投与の安全性と有効性を検討する多施設第2相臨床試験を行った。
患者は経口エトポシド(60mg/m2/day)を1〜10日まで投与し、その後11日間は休薬するスケジュールで治療を行った。
中国の10カ所の病院から参加した75例の転移性乳がん患者を対象にした。7例(9.3%)が部分奏功、29例(38.7%)が病状安定を示した。9例(12%)は24週間以上の病状安定で、臨床的有効性は21.3%(16/75)であった。
無増悪生存期間の中央値は4.5ヶ月(1.3〜7.7ヶ月)であった。
本臨床試験を受ける前に3種類以上の処方で抗がん剤治療を受けていた38例では、2例(5.3%)が部分奏功、3例(7.9%)が24週以上の病状安定、臨床的有効性は13.2%であった。
グレード3から4の有害事象は白血球減少(13.3%)、好中球減少(17.9%)、貧血(2.7%)、嘔吐(2.6%)、脱毛(1.3%)であった。
以上より、経口エトポシドは高度に治療を受けている転移性乳がんの患者に対して有効で安全性の高い治療と言える。
同様の臨床試験はイタリアからも報告されています。
A retrospective analysis of the activity and safety of oral Etoposide in heavily pretreated metastatic breast cancer patients.(高度に抗がん剤治療を受けた転移性乳がん患者における経口エトポシド治療の有効性と安全性に関する後ろ向き解析)Breast J. 2015 May-Jun;21(3):241-5. doi: 10.1111/tbj.12398. Epub 2015 Mar 15.
【要旨】
転移を認める乳がんは抗がん剤治療が主体になるが、複数の処方による抗がん剤治療行われ効果が無くなってくると治療は極めて困難になる。経口エトポシドは転移性乳がんの治療に有効であることが臨床試験で示されている。
しかしながら、多くの新薬の開発や、エトポシドの毒性が過大に認識されているためか、エトポシドの使用は減少している。
本研究では、現在多く使用されている抗がん剤治療の複数のレジメンで高度に治療を受け、効果が無くなった転移性乳がんの患者を対象にして、経口エトポシドの臨床的有用性と安全性について検討した。
本研究の対象は66例の転移性乳がんの患者で、すでに複数のレジメン(regimen)で抗がん剤治療を受けており、そのレジメンの数の中央値は8(2〜13)であった。
患者は1日50mgのエトポシドを20日間連続で経口で摂取し、ついで7日間の休薬期間を置いた。
無増悪生存期間の中央値は4ヶ月、奏功率は4%、臨床的有効性は18%、治療開始からの全生存期間は11ヶ月であった。
臨床的に重度の毒性はほとんど認めなかった。エトポシドの副作用によって治療を中断した患者はいなかった。
その臨床的有効性と低毒性と低価格という特徴から、高度に抗がん剤治療を受けた転移性乳がん患者の治療に経口エトポシドは有益な選択肢であることが示唆された。
同様の目的でシクロフォスファミドとエトポシドの併用による治療も行われています。
Metronomic oral chemotherapy with old agents in patients with heavily treated metastatic breast cancer.(高度に治療を受けた転移性乳がん患者における古い経口抗がん剤によるメトロノミック・ケモテラピー)J Cancer Res Ther. 2015 Apr-Jun;11(2):287-90. doi: 10.4103/0973-1482.154008.
【要旨】
研究の背景:高度に抗がん剤治療を受けた転移性乳がんの患者に対するシクロフォスファミドとエトポシドのメトロノミック投与の有効性を検討した。
方法:転移を有する乳がん患者77例を対象に、シクロフォスファミド(50mg/日の連続投与)とエトポシド(2 x 50mg/日、週2回)の投与を行った。対象の患者はアントラサイクリン系、タキサン系、代謝拮抗剤の抗がん剤で治療を受け、効果が無くなった症例であった。
結果:無増悪生存期間の中央値は7.03ヶ月(5.06〜8.99ヶ月)、全生存期間の中央値は32.5ヶ月(22.5〜42.4ヶ月)であった。
結論:高度に治療を受けている転移性乳がんの患者に治療に、シクロフォスファミドとエトポシドによるメトロノミック・ケモテラピーは新規で有効性の高い治療法となりうる。この治療法は毒性が低く、安価に実施できる。生存期間の延長と低価格ということから非常に有益な治療法となりうる。
以上のようなシクロフォスファミドやエトポシドの低用量投与や間歇的投与と、樹状細胞やナチュラルキラー細胞や抗原特異的キラーT細胞の活性を高めて、抗腫瘍免疫を積極的に高める治療法を併用するとより効果が期待できそうに思います。
例えば次のような治療法が考えられます。
① シクロフォスファミドを低用量(50mg/日または週3〜4回)とエトポシド(50mg/日を1ヶ月のうち最初の5〜7日間)を投与してがん細胞に免疫原性細胞死を誘導する。
② IL-12の産生を増やし、樹状細胞の成熟を促進するためにピドチモドを1日400〜800mgを服用。
③ ナイーブヘルパーT細胞のTh1への分化を抑制し、骨髄由来抑制細胞を誘導するプロスタグランジンE2の産生を抑制するCOX-2阻害剤のセレコックスを1日200〜400mg服用(朝と夕の食後)
④ 骨髄由来抑制細胞を抑制し、抗腫瘍免疫を増強するシメチジンを1日800mg服用する。
⑤ T細胞からTh1サイトカイン産生を増やし、キラーT細胞やナチュラルキラー細胞の働きを高める漢方薬(特に紅参、黄耆、川芎などを多く使用)を服用する。
⑥ さらに、オプションとして骨髄由来抑制細胞を分化誘導して抑制活性を減らすレチノイド(イソトレチノイン 10〜20mg/日)とビタミンD3(2000〜4000 IU/日)を併用する。
⑦ 皮膚転移や表層部のリンパ節があるときはイミキモドクリームを病変近くに塗布する。
抗腫瘍免疫の増強法に関する詳細はこちらへ:
| « 651)駆虫薬メ... | 653)免疫原性... » |