がんの予防や治療における漢方治療の存在意義を考察しています。がん治療に役立つ情報も紹介しています。
「漢方がん治療」を考える
カレンダー
| 2025年4月 | ||||||||
| 日 | 月 | 火 | 水 | 木 | 金 | 土 | ||
| 1 | 2 | 3 | 4 | 5 | ||||
| 6 | 7 | 8 | 9 | 10 | 11 | 12 | ||
| 13 | 14 | 15 | 16 | 17 | 18 | 19 | ||
| 20 | 21 | 22 | 23 | 24 | 25 | 26 | ||
| 27 | 28 | 29 | 30 | |||||
|
||||||||
goo ブログ
過去の記事
カテゴリ
最新の投稿
最新のコメント
最新のトラックバック
ブックマーク
プロフィール
検索
gooおすすめリンク




| URLをメールで送信する | |
| (for PC & MOBILE) | |
875)抗がん剤の臨床試験の結果はどの程度信頼できるのか
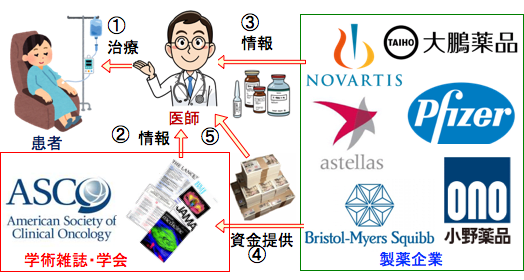
図:医師は薬を使って患者を治療する(①)。医師は医薬品や治療法の情報を学術雑誌や学会から得ている(②)。製薬会社主催の説明会や講演会などからも医薬品の情報を得ているが、製薬企業は販売促進に有利な情報を与える傾向にある(③)。学術雑誌には広告の名目で多額の資金が製薬企業から入っている。医学雑誌の編集委員医師にも製薬会社から金銭が渡っている。学会も製薬企業の資金が入っており、製薬企業の思惑に沿った内容の論文やガイドラインが作成されることも多い(④)。医師(謝礼)や大学(研究費)にも製薬企業の資金は流入している(⑤)。製薬企業は利益追求のために販売促進を積極的に行っているので、大して効かない高額な抗がん剤が多く使われている。
875)抗がん剤の臨床試験の結果はどの程度信頼できるのか
【報告されている臨床試験の結果が正しいとは限らない】
研究者が学術雑誌に投稿した論文は査読によって審査され、掲載の可否が決められます。査読(peer review;ピア・レビュー)というのは、同分野の専門家による評価や検証のことです。
査読論文とは、ピアレビューされた論文のことで、ピア(peer)は同業の審査者を意味し、レフリーともよばれます。投稿論文を受け取ると、編集事務局は1〜3人程度のレフリーに論文の審査を依頼します。
レフリーは、実験方法は適切であるか、統計処理は正しいか、研究の目的と結果と結論に整合性があるか、などをチェックして、掲載の可否についての意見を添えて事務局に送り返します。
しかし、レフリーが論文に掲載された実験を再現するわけでは無いので、データの捏造や改ざんがあっても見抜く事はできません。
つまり、査読論文でも、超一流の学術雑誌でも、実験自体をやっていないようなでたらめな論文でも掲載されています。そのような事件(論文捏造)は今まで数多く起こっています。
では、再現性の無い研究論文はどの程度あるかというと、10%とか20%程度という報告もありますが、半数以上というのが一般的な意見です。つまり、論文で報告された実験結果の半分以上は真実で無い(再現性がない)ということです。
基礎研究の論文に再現性があるかどうかは医薬品開発で重要なので、製薬企業では重要な論文については追試を行って再現性を確かめています。その結果が報告されています。
一つはバイエルヘルスケア社(Bayer HealthCare)の研究所からの報告です。
Believe it or not: how much can we rely on published data on potential drug targets?(信じるか信じないか:医薬品のターゲットに関する発表された研究データはどの程度信頼できるのか?) Nat Rev Drug Discov. 2011 Aug 31;10(9):712. doi: 10.1038/nrd3439-c1.
この報告では、社内で重要と考えたがん領域の論文を研究所内で再実験して追試を試みたところ、研究論文の約25%しか再現性がありませんでした。
アムジェン社からの内部資料による報告もあります。Natureの論文ですので、かなりの問題提起になっています。
Drug development: Raise standards for preclinical cancer research(医薬品開発:前臨床がん研究の基準を高める)Nature. 2012 Mar 28;483(7391):531-3.
この報告では過去10年にわたって発表されたがん研究で高い評価を受けた53の論文について、アムジェン社の血液腫瘍学部門の研究者が再現実験を行った内部資料が公開されています。この53編の半分は一流学術誌の論文で、引用がすでに平均230を超える論文で、この分野への影響の高い論文が選ばれ、再現性が検討されています。
結果はバイエル社の結果よりさらに悪く、53編中の6編(11%)だけが再現可能だという結果でした。
これらの再現性のない基礎研究の結果を元に、一連の臨床試験が開始され、多くの患者が意味の無い試験を受けていることになります。
つまり、がん治療の分野で臨床試験の失敗が多いのは、公開された基礎研究の質が悪いからだということが推定されています。
がん治療薬の開発は、がんの生物学と新しいターゲット分子に関する文献に大きく依存しているのですが、その文献の再現性が極めて乏しい状況だから臨床試験で失敗しているという指摘です。
Natureの論文は「腫瘍と患者の両方の不均一性から生じる臨床試験の厳しさと課題に耐えるために、前臨床試験の結果は非常に頑強でなければならない」「基礎研究や前臨床試験の再現性を高めるように基準を高めなければならない」「科学的プロセスには、最高水準の品質、倫理、厳格さが求められる」などと言っています。
論文で発表された実験結果の再現性の欠如にはいくつかの理由が考えられます。
新規で肯定的な結果ほど学術雑誌に掲載されやすいので、論文数で業績を評価される研究者は、無理してデータを良く見せる可能性もあります。そのため、統計分析でデータを少しだけ改ざんする場合もあります。
統計的有意差を示すときに「p値」が使われます。P値は確率(probability )のことで、「p<0.05」は「確率が5%未満である」ということを意味しています。
「この差が、単なる偶然で生じた誤差である危険率が5%未満」ということを意味しています。
しかし、これは、有意な差が無い確率が5%あるということでもあります。通常、p<0.05であれば、統計的な有意差があると評価されます。
最近、P値の閾値は0.05から0.005に引き下げるべきであると、統計学の大家たちは主張しています。Nature (2017-08-03) | doi: 10.1038/nature.2017.22375 | Big names in statistics want to shake up much-maligned P value
現在の科学は「再現性の危機」に苦しんでいて、研究者も助成機関も出版社も、学術文献は信頼できない結果にまみれているのではないかと不安を募らせています。それで、72人の著名な研究者が、新たな発見をしたと主張する際の証拠の統計的基準の低さが再現性の危機の一因になっているとする論文を発表しました。
科学の多くの分野で、新発見を主張する際の実証結果に関する統計的基準があまりに低いという指摘です。p値が0.05より低い結果を「統計的に有意な」発見とすることは、実験や手続きや報告についての他の問題が無い場合でも、偽陽性を高い割合でもたらすことになるので、新発見の統計的有意性の閾値がp<0.05と定義されている分野について、それをp<0.005に変更するよう提言しています。
【多くの抗がん剤が1回の臨床試験の結果だけで承認されている】
有効な薬が少ないがん治療の現場に新薬を早く届けるために、医薬品の承認を迅速化することは意味があります。
米国食品医薬品局(FDA)は 1992 年に加速承認規制( Accelerated Approval regulations)を制定しました。
「Accelerated Approval regulations」は、特定の医薬品や治療法が早期に承認を受けることができるようにするための規制やプロセスのことを指します。
具体的には、通常の臨床試験プロセスを経ての承認を待つ代わりに、より早期の段階や予備的なデータに基づいて、条件付きで承認されることがあります。
抗がん剤の場合、生存期間の延長や生活の質(QOL)の改善が真のエンドポイントになりますが、生存期間の延長を証明するには時間がかかるので、より短期間に評価できる代用エンドポイント(奏功率や無増悪生存期間)の結果で承認が可能になりました。
「がんが縮小すれば延命やQOL改善につながるだろう」「無増悪生存期間が延長すれば全生存期間も延長するだろう」という予測に基づいています。
さらに、迅速承認では1回の臨床試験の結果で承認されていることも多くあります。承認を急ぐのが目的なので、再現性を確認するための別の臨床試験の実施は求めないということです。
以下のような報告があります。
Single pivotal trials with few corroborating characteristics were used for FDA approval of cancer therapies(がん治療薬のFDA承認には、裏付けとなる特徴がほとんどない単一の主要な試験が使用された)J Clin Epidemiol. 2019 Oct;114:49-59.
新しいがん治療法は、単一の主要な試験のみからの証拠によって承認されることが多いのですが、その証拠の信頼性には懸念があります。
この論文では2000 年から 2016 年の間に、がんに対する新薬および生物学的製剤の FDA(米国食品医薬品局) 承認を裏付けるすべての臨床試験のメタ疫学的評価を行っています。
100 件の承認のうち 35 件(20 件のランダム化比較試験と 15 件の単群試験)は、有益な効果に関するさらなる裏付けのない、単一の試験からの証拠に基づいていました。
承認数は、2006 年以前は 1 件でしたが、2011 年以降は 23 件に大幅に増加していました。
1回の臨床試験の結果で承認された場合、その結果が再現性のある結果なのかどうかは、承認された時点では不明です。代用エンドポイントの奏功率(がんの縮小率)や無増悪生存期間と全生存期間との相関は低いことも明らかになっています。つまり、奏功率が高く無増悪生存期間が延長しても、全生存期間が延長しないことは多くあります。がんが縮小しても生活の質(QOL)が良くならないことも多く、むしろ副作用でQOLが悪化することも多くあります。
迅速承認(Accelerated Approval)の考え方は、患者にとっては新しい治療法を早期に利用できるメリットがあります。製薬メーカーにとっても、早く承認されれば、特許期間が長く残るので、利益につながります。
しかし、十分なデータが揃っていない段階での承認となるため、安全性や有効性に関する懸念も伴います。
最近の論文でも、ランダム化比較試験の結果がなくても承認されている抗がん剤が多いことが明らかになっています。その現状を心配する意見があります。以下のような報告があります。
The evidence base of US Food and Drug Administration approvals of novel cancer therapies from 2000 to 2020. (2000 年から2020 年までの米国食品医薬品局の新規がん治療薬承認のエビデンス) Int J Cancer. 2023 Jun 15;152(12):2474-2484.
【要旨の抜粋】
医薬品を承認する米国食品医薬品局(FDA)が抗がん剤の承認を加速する方針であるため、抗がん剤治療による生存と生活の質(QoL)に対する利益と害について十分なエビデンスが無い段階で承認されている可能性があるとの懸念が指摘されている。
2000 年から 2020 年の間に 米国食品医薬品局(FDA)によって初めて承認されたすべての抗がん剤に関する臨床試験を分析した。
2000年から2020年の間に、FDAは190の臨床試験に基づいて、156の適応症に対して145の新規抗がん剤を承認した。適応症の半分(49%)はランダム化比較試験の証拠なしに承認され、82% は 1 回の臨床試験のみの実施であった。
全生存期間は試験の14% で主要評価項目であり、QoL(生活の質) のデータは 25% から入手可能であった。全生存期間の延長の中央値は 2.55 か月であった。QoL の改善は 156 件中 7 件 (4%) で報告された。
最近になるほど、優先審査(priority review)の使用が増加し、適応ごとの平均試験数は 1.45 から 1.12 に減少した。
この21 年間、新しい抗がん剤は通常、代替エンドポイントを用いた対照群なしの、しばしば管理されていない単一の臨床試験に基づいて承認されてきた。新薬が生存率や QoL を改善するという確かな証拠がなくがん患者に使用され、改善に向けた兆候も認められない。
過去 21 年間に、FDA は 156 のがん治療適応症に対して 145 の新薬を承認しました。大部分は優先審査を受け、約半数は迅速承認を受けました。
承認5件中4件は、単一試験からの証拠のみに基づいており、2例に1件はランダム化比較試験ではなく、全生存期間以外の代用エンドポイント、つまり無増悪生存期間または腫瘍縮小(奏功率)に基づいていました。
治験は通常小規模で、多くは患者数 250 人未満でした。対照群を使用しないほぼすべての試験では、奏功率が主要評価項目でした。
つまり、がんが縮小すれば、抗がん剤として承認するという方針です。腫瘍の縮小率(奏功率)は全生存期間と相関しないことは常識ですが、腫瘍が縮小すれば抗がん剤として承認するというのがFDAの方針のようです。
ランダム化比較試験では、全生存期間が主要評価項目となったのは 28% のみで、最も頻度の高い主要評価項目は無増悪生存期間(51%) でした。
このような状況は何年も批判されてきましたが、過去 20 年間、新規抗がん剤の試験において全生存期間を主要評価項目に使用する傾向や、 ランダム化比較試験をより頻繁に利用する傾向は観察できなかったという結果でした。
様々な薬が大量に承認されているにもかかわらず、それらの薬が患者にもたらすメリットはさほど大きくなく、絶対的な利益は全生存期間が2ヶ月から4ヶ月間延長された程度でした。
ランダム化比較試験でQoLを改善することが示された抗がん剤はごく少数でした(145件中7件、5%)。
簡単にまとめると、「最近の抗がん剤の新薬の臨床試験は、承認のスピードを早めるために、再現性や信頼性が保証されていない」ということです。
【迅速承認後に有効性が否定されて撤回される抗がん剤は少なくない】
有効な治療法がないがん治療の臨床現場に、速く新薬を届けたいという目的で迅速承認が認められています。しかし、迅速承認で使用された抗がん剤の中に市販後の調査で有効性が否定され、市場から撤回される抗がん剤もあります。その数は少なくありません。20%も撤回されていると報告されています。以下のような論文があります。
Withdrawn accelerated approvals for cancer indications in the USA: what is the marketing authorisation status in the EU?(米国におけるがん適応症の早期承認の撤回: EU における販売承認状況はどうなっていますか?)Lancet Oncol. 2023 Sep;24(9):e385-e394.
【要旨】
2023年4月の時点で、1992年以来米国食品医薬品局(FDA)が認めたがん適応症の早期承認のうち23件が米国市場から撤回され、そのうち17件(74%)が過去3年間に撤回された。
EU におけるこれらの適応症の販売承認状況は報告されていない。FDA と欧州医薬品庁 (EMA) からの関連文書のレビューは、FDA によって取り消されたがん適応症の早期承認が現在までに EU で販売承認されているかどうかを調査し、これらの適応症の承認履歴を比較するために行われた。
2023年4月20日の時点で、米国でがん適応症の早期承認を撤回した23件中9件(39%)が、EUで同様の適応症の販売承認を取得していることが判明した。比較すると、EUから取り消されたがん適応症の条件付き販売承認は2件のみで、どちらも米国では承認されていない。
これらの調査結果は、FDA と EMA の間の承認方針に相違があることを示しており、米国の一部の患者グループが適切な医療を受けられていないか、EU の一部の患者グループがプラスの効果のない薬で治療されている可能性を示唆している。
FDA の迅速承認規制と同様に、欧州医薬品庁 (EMA) は 2006 年に条件付き販売承認を確立しました。
この論文は、医薬品の規制機関である米国の食品医薬品局(FDA)とEU(欧州連合)の欧州医薬品庁(EMA)が、迅速承認した抗がん剤に関して緊密に情報交換するべきだと提言しています。
1992年12月11日から2023年4月20日までに、FDAが早期承認したがん治療適応114件のうち23件(20%)が米国市場から撤回され、そのうち17件(74%)は過去3年間に米国市場から撤回されました。
米国食品医薬品局(FDA)が有効性が証明されないと判断したがん治療薬23個のうち9個はEUでまだ使用されているということです。つまり、EUでがん治療に使われている薬のうち少なくとも9個は効果のないものが使用されているという事実です。
FDA の迅速承認規制は、主に 2 つの理由から批判されています。第一に、臨床試験の大部分で代用エンドポイントが使用されています。そのため、治験実施計画書で指定された主要評価項目を達成したとしても、薬剤の臨床上の利点には依然として疑問が残る可能性があります。代用エンドポイント(奏功率や無増悪生存期間)と真のエンドポイント(全生存期間やQOL改善)の創刊が低いからです。
第二に、承認後に行われた確認試験で有効性が示されなかった薬が、その適応症が取り消されるまで何年も薬剤のラベルと臨床診療ガイドラインに記載されたままでした。
1992年12月11日から2023年4月20日までに、FDAによってがん適応症の早期承認が与えられ、その後米国市場から早期承認が取り消された医薬品とその適応症のリストは以下です。
Atezolizumab(アテゾリズマブ):抗PD-L1抗体医薬品。商品名はテセントリク(Tecentriq)
Triple negative breast cancer(トリプルネガティブ乳がん)
First-line urothelial carcinoma (尿路上皮癌の第一次治療)
Second-line urothelial carcinoma(尿路上皮癌の第二次治療)
Belantamab mafodotin(ランタマブ マホドチン):抗BCMA抗体薬物複合体
Multiple myeloma(多発性骨髄腫)
Bevacizumab(ベバシズマブ):血管内皮細胞増殖因子に対するモノクローナル抗体。商品名はアバスチン
Breast cancer(乳がん)
Celecoxib(セレコキシブ):COX-2阻害剤
Familial adenomatous polyposis(家族性腺腫性ポリポーシス)
Durvalumab(デュルバルマブ):抗ヒトPD-L1モノクローナル抗体
Urothelial carcinoma(尿路上皮がん)
Duvelisib(デュベリシブ):PI3キナーゼ阻害剤
Follicular lymphoma (濾胞性リンパ腫)
Fludarabine(フルダラビン):ヌクレオシド系抗腫瘍性代謝拮抗剤
Chronic lymphocytic leukaemia(慢性リンパ性白血病)
Gefitinib(ゲフィチニブ):上皮成長因子受容体(EGFR)チロシンキナーゼ阻害剤
Non-small cell lung cancer(非小細胞肺がん)
Gemtuzumab ozogamicin(ゲムツズマブ オゾガマイシン):抗体薬物複合体。(CD33に対するヒト化モノクローナル抗体部分と、細胞毒性を有するカリケアミシン系のオゾガマイシン部分から成る)
Acute myeloid leukaemia(急性骨髄性白血病)
Idelalisib(イデラリブ):ホスホイノシチド 3-キナーゼ(PI3K)阻害剤
Follicular lymphoma (濾胞性リンパ腫)
Nivolumab(ニボルマブ): ヒト型抗ヒトPD-1モノクローナル抗体医薬品。商品名はオプジーボ
Small-cell lung cancer(小細胞肺がん)
Hepatocellular carcinoma(肝細胞がん)
Olaratumab(オララツマブ):PDGFR-α活性化阻止抗体
Sarcoma(肉腫)
Panobinostat(パノビノスタット):非選択的ヒストン脱アセチル化酵素阻害薬
Multiple myeloma(多発性骨髄腫)
Pembrolizumab(ペンブロリズマブ):ヒト化抗ヒトPD-1モノクローナル抗体。商品名はキイトルーダ
Small-cell lung cancer(小細胞肺がん)
Gastro-oesophageal cancer (胃食道がん)
Romidepsin(ロミデプシン):ヒストン脱アセチル化酵素(HDAC)阻害剤
T-cell lymphoma(T細胞リンパ腫)
Tositumomab and [131I]iodine tositumomab(トシツモマブと[131[I]ヨウ素 トシツモマブ):CD20抗原を標的とするマウスモノクローナル抗体にヨウ素-131を結合した放射性医薬品
Non-Hodgkin lymphoma(非ホジキンリンパ腫)
Umbralisib(ウンブラリシブ):PI3K-δおよびカゼインキナーゼ-1(CK1)-ε阻害薬
Marginal zone lymphoma(辺縁帯リンパ腫)
Follicular lymphoma (濾胞性リンパ腫)
Vincristine(ビンクリスチン):植物アルカロイド。微小管阻害剤
Acute lymphocytic leukaemia(急性骨髄性白血病)
例えば、FDAは2016年10月に軟部肉腫治療薬としてEli Lilly社のolaratumab(オララツマブ)が迅速承認されました。
オララツマブは、血小板由来増殖因子 (PDGF) 受容体アルファを阻害する抗体です。PDGF 受容体が刺激されると、腫瘍の増殖が引き起こされます。オララツマブはこれらの受容体をブロックすることで作用し、腫瘍の増殖を遅らせたり停止したりするのに役立つ可能性があります。
転移性軟部肉腫患者 133 人を対象としたランダム化臨床試験が行われ、生存期間中央値は、ドキソルビシン単独投与の患者の14.7カ月と比較して、ドキソルビシン+オララツマブ併用群は26.5カ月で、統計的に有意に延長する結果が得られたので、FDAは迅速承認しました。
しかし、承認後に実施された第3相試験で、ドキソルビシン単独投与群とドキソルビシン+オララツマブ併用群の生存期間に差がつかなかったため、イーライ・リリー社は2019年1月に販売を中止しています。
予備的な臨床試験で良好な結果が得られましたが、その有効性が再現できなかったために、承認が撤回された例です。
米国食品医薬品局(FDA)は、医薬品の市場投入を加速し、安全性と有効性を確保するという競合する圧力にさらされています。一方では、患者、家族、支援者らは、重篤で生命を脅かす病気の治療法となる新薬の迅速な承認を求めています。彼らは遅れが潜在的に致命的であると信じています。
製薬会社も同様に、新薬をできるだけ早く市場に投入して収益を上げ始めたいと考えています。
その一方で、医師や監視団体は、サリドマイド、ロフェコキシブ、ベバシズマブ、その他の無効または有害な薬物の経験を思い出し、安全性、実際の有効性、そして安全性と有効性が証明されていない薬物の使用に伴う経済的無駄を懸念しています。
このような多方面からの圧力によって、「科学的エビデンスに基づいた臨床試験」の実施が困難になっているのかもしれません。つまり、有用性の高い治療薬が出てこないことへの焦りが、規制機関と患者と医師と製薬メーカーを間違った方向に導いているようです。
その結果、無益で有害な薬が癌患者に使われています。つまり、標準治療の抗がん剤治療のエビデンスレベルは極めて低いと言えます。
【統計的に有意差があっても、臨床的に意味のある有益性の無い抗がん剤が多い】
以上のような理由によって、「臨床的に意味のある有益性」に乏しいがん治療薬が量産されています。
ある新規のがん治療薬を使った場合の生存期間が、プラセボ(偽薬)群あるいは既存のがん治療薬の生存期間に比べて、統計的有意に勝っていれば、その新しいがん治療薬は新しい治療法として認められます。
しかし、生存期間の延長で統計的に有意差を示しても、臨床的に意味のある有益性があるとは限りません。
例えば、生存期間が12ヶ月から14ヶ月に延長しても、強い副作用を伴い、生活の質が著しく低下するような治療薬であれば、臨床的に意味がある有益性があるとは言えないと思います。
固形腫瘍に使用されている71の薬物による生存期間の延長の中央値はわずか2.1ヶ月という報告もあります。(JAMA Otolaryngol Head Neck Surg 2014;140:1225–36.)
がん治療薬のわずかな利益も、平均的な患者集団より若年で合併症の少ない患者において実施される臨床試験で認められるだけという指摘もあります。患者全体を対象にすると、抗がん剤のメリットとデメリットの微妙なバランスの中で、わずかな利益は完全に消滅する可能性があるのです。
前述のように、多くのがん治療薬が迅速承認されていますが、患者の生存を改善する十分な証拠を得て市場に参入する抗がん剤はほとんど無いのが実情です。有益性が認められた場合でも、その利益はわずかであるため、異なる病状の患者集団を対象にした場合、その利益は失われる可能性があるのです。つまり、がん以外に病気をもっていない元気ながん患者を対象に行われた臨床試験の結果は、併存疾患をもつ高齢者には当てはまりません。
欧州臨床腫瘍学会(European Society for Medical Oncology、ESMO)はがん治療法を評価するツールとして臨床的ベネフィット・スケール・マグニチュード(Magnitude of Clinical Benefit Scale:MCBS)を発表しています。
ESMO-MCBSは、がん治療薬の治療効果を評価するために設計されており、有効性(全生存期間と無増悪生存期間の絶対的な増加およびハザード比の95%信頼区間の下限)と、生活の質(QOL)または毒性をそれぞれ検討します。新規治療法のデータは、病状ごとの予後(対照群での治療奏効期間または生存期間)に関して分析されて臨床的利益が評価されます。
例えば、根治(cure)を目指す治療(手術前の抗がん剤治療や手術後の補助化学療法)では、3年以上の追跡で何%の生存率の増加があるかで有効性の程度が評価できます。
進行がんの緩和的化学療法では、対照群が12ヶ月以下の生存期間の場合、3ヶ月以上の生存期間の延長があれば臨床的に意味がある有用性があると言えます。対照群が12ヶ月以上の生存期間の場合、臨床的に意味があるというには5ヶ月以上の延命が必要かもしれません。
このように、病気の進行状況に応じて、生存期間の延長や生活の質の改善や副作用(毒性)を評価して、臨床的に意味のある有用性を評価するツールがESMO-MCBSです。
このESMO-MCBSの評価法を使って、最近承認われた抗がん剤やランダム化比較試験を検証すると、臨床的に意味のある有用性を示した抗がん剤は2割以下のようです。
例えば、以下のような報告があります。
Do Contemporary Randomized Controlled Trials Meet ESMO Thresholds for Meaningful Clinical Benefit?(最近の無作為化比較試験は意味のある臨床的利益のためのESMO閾値を満たしているのか?)Ann Oncol. 2017 Jan 1;28(1):157-162.
【要旨】
背景:欧州臨床腫瘍学会(European Society for Medical Oncology;ESMO)は、固形腫瘍に対する化学療法の有効性を評価するツールとして臨床的ベネフィット・スケール・マグニチュード(ESMO Magnitude of Clinical Benefit Scale:ESMO-MCBS)を最近発表した。この研究では、最近報告されているランダム化比較試験がESMO-MCBSで評価される意味のある臨床的有益性の閾値に達しているかどうかを評価した。
方法:2011年から2015年の間に論文に公表された乳がん、非小細胞性肺がん、結腸直腸がん、膵臓がんに対する化学療法の有効性を検討したランダム化比較試験を解析した。
臨床試験の特徴と結果に関するデータを抽出し、これらのデータをESMO-MCBSで評価した。個々の臨床試験が、ESMO-MCBSによって定義された臨床的有益性を評価できるような試験デザインであるかどうかも検討した。
結果:対象となるランダム化比較試験は277件(乳がん40%、非小細胞性肺がん 31%、結腸直腸がん22%、膵臓がん6%)であった。サンプルサイズ(対象になった人数)の中央値は532で、83%は製薬企業からの資金提供を受けていた。
277件のランダム化比較試験の中で、138件(50%)の試験で治療群の有効性は対照群より統計的有意であった。これら有効の結果が得られた試験のわずか31%(43/138)の結果がESMO-MCBSによる臨床的に意味のある利益閾値を満たした。
治癒的意図を有する治療のランダム化比較試験(RCTs with curative intent)では、有効性を示した31件中19件(61%)で意味のある臨床的有益性の閾値を満たしていた。一方、緩和目的の抗がん剤治療のランダム化比較試験では、統計的有意な有効性を示した107件中24件(22%)が臨床的に意味のある有益性の閾値を満たしていた。
ESMO-MCBSが適用され得る226件のランダム化比較試験のうち、ESMO-MCBSの有益性の閾値を満たすことができる臨床試験のデザインで臨床試験を行っていたのは31%(70/226)であった。
結論:統計的に有意な有効性を示した最近のランダム化比較試験のうち、欧州臨床腫瘍学会による臨床的ベネフィット・スケール・マグニチュード(ESMO-MCBS)の基準で臨床的に意味のある有益性(meaningful clinical benefit)の閾値に達したのは3分の1以下であった。これは全ての公開された試験の15%に過ぎない。
研究者や資金提供機関や規制機関や製薬業界は、今後のランダム化臨床試験の設計において、意味のある臨床的利益のために、より厳しい基準を採用すべきである。
この論文の結果は下図にまとめています。

図:(左)欧州臨床腫瘍学会(European Society for Medical Oncology、ESMO)はがん治療法を評価するツールとして臨床的ベネフィット・スケール・マグニチュード(Magnitude of Clinical Benefit Scale:MCBS)を発表している。ESMO-MCBSは、がん治療薬の治療効果を評価するために設計されており、全生存期間と無増悪生存期間の延長、生活の質(QOL)または毒性をそれぞれ検討し、病状ごとの予後(対照群での生存期間または治療奏効期間)に分類して分析され、臨床的有益性を総合評価する。費用(コスト)は考慮されない。
(右)2011年から2015年の間に論文に公表された乳がん、非小細胞性肺がん、結腸直腸がん、膵臓がんに対する化学療法の有効性を検討したランダム化比較試験は277件で、このうち138件(50%)の試験で治療群は対照群より統計的有意な有効性を示した。これら有効な結果が得られた試験138件中でESMO-MCBSによる臨床的に意味のある利益閾値を満たしたのは43件であった。これは公開された全ての試験の15.5%に過ぎない。(出典:Ann Oncol. 2017 Jan 1;28(1):157-162.)
別の研究グループからも同様の調査結果が報告されています。
2011から2016年に欧州医薬品庁からに承認を受けた38種類のがん治療薬に対する70件の臨床試験を、欧州臨床腫瘍学会の臨床利益スケール(ESMO-MCBS)で評価しています。
Five years of EMA-approved systemic cancer therapies for solid tumours-a comparison of two thresholds for meaningful clinical benefit. (固形腫瘍のための欧州医薬品庁承認の全身がん治療法の5年間 – 意味のある臨床的利益のための2つの閾値の比較。)Eur J Cancer. 2017 Sep;82:66-71.
前述のようにESMO-MCBSは、がん治療薬の治療効果のレベルを評価するために設計されています。最初に発表されたESMO-MCBSとその改良版によって定義された「意味のある臨床的利益」の閾値を満たすものがどの程度存在するかを検討しました。
その結果、「意味のある臨床的利益」があると評価されたがん治療薬は、最初のESMO-MCBSの基準では21%、改良版の基準では11%しかありませんでした。つまり、基準の違いによって評価は変わりますが、承認されたがん治療薬の80〜90%は臨床的に意味のある有益性を示していないという結果です。
ランダム化臨床試験で統計的有意差があっても、臨床的に意味のある有益性が証明されている抗がん剤は3分の1程度です。統計的有意に生存期間が延長しても、それが強い副作用を伴って3ヶ月程度であれば、その治療を受ける意味があるのか、患者さんも医師も考える必要があると思います。
ESMO-MCBSは治療にかかる費用(コスト)は考慮していません。国によって費用負担が異なるので評価に加えられないためです。しかし、最近のがん治療薬の平均は1ヶ月に100万円を超えています。コストを考慮すると、有益性はもっと低下するかもしれません。
【製薬企業がスポンサーの臨床試験は肯定的な結論を出す傾向にある】
治験や臨床試験において、医師あるいは製薬企業などが新薬承認申請などに自分たちの都合の良いように症例データを書き換えたりすることが以前から起こっています。
製薬企業がスポンサー(資金提供)の臨床試験は肯定的な結論を出す傾向にあることが報告されています。
以下の報告はコクラン・コラボレーション(The Cochrane Collaboration)によるレビューです。
Industry sponsorship and research outcome.(企業のスポンサーシップと研究の結果)Cochrane Database Syst Rev. 2017 Feb 16;2:MR000033. doi: 10.1002/14651858.MR000033.pub3.
【要旨の抜粋】
研究の背景:医師が医療を行う際に影響を及ぼす臨床研究は、医薬品や医療機器を製造している企業による資金提供が益々増加している。以前の系統的レビューでは、製薬産業が資金提供者(スポンサー)となった研究は、他のスポンサーシップの研究と比較して、資金提供企業の製品により好都合な結果を出す傾向が高いことが判明した。このレビューは、以前のCochraneレビューの更新版であり、スポンサーシップと研究成果の関連性に関する実証的研究を含んでいる。
目的:業界がスポンサーとなった医薬品と医療機器の研究が、他のスポンサーシップの研究と比較してより好ましい結果をもたらし、バイアス(偏り)のリスクが異なるかどうかを調査する。
調査方法:今回のアップデート(更新)では、MEDLINE(2010年〜2010年2月)、Embase(2010年〜2015年2月)、Cochrane Methodology Register(2015年、第2版)、Web of Science(2015年6月)を検索した。加えて、検索した論文中の引用文献リスト、以前の体系的レビュー、著者ファイルを検索した。
選択基準:業界からの資金提供による医薬品または医療機器の臨床研究と、他のスポンサーシップによる臨床研究とを定量的に比較した横断的研究、コホート研究、体系的レビューおよびメタアナリシスを選択した。言語制限は行わなかった。
データ収集と解析:2人の査定者が抄録をスクリーニングし、関連する論文を特定して解析に含めた。2人の査定者がデータを抽出し、追加の未発表データについては論文の著者に問い合わせた。
2人の査定者は、解析した論文の偏見のリスクを評価した。二分法データ(95%信頼区間を有する)に関するプールされたリスク比(RR)を計算した。
主な結果:このアップデートには27の新しい論文が含まれ、この系統的レビューには合計75の論文が含まれている。 企業以外の資金提供による臨床研究に比べて、企業がスポンサーになっている臨床研究では、有効性がより高く(RR:1.27(95%CI:1.17〜1.37))、有害性のデータも好ましいもので(RR:1.37(95%CI:0.64〜2.93))、より好意的な結論(RR:1.34(95%CI:1.19-1.51))であった。
スポンサーシップと結論の関連性において薬物と医療機器の研究の間に差は見られなかった。
業界が資金提供する臨床研究では、企業から支援されていない研究と比較して、結果と結論の一致がより少なかった(RR:0.83(95%CI:0.70-0.98))。
著者の結論:製造会社による薬物および医療機器の臨床研究のスポンサーシップ(資金提供)は、他の資金源によるスポンサーシップよりも、より好ましい有効性の結果および結論につながる。私たちの分析は、標準的な「バイアスのリスク」評価では説明できない業界バイアス(industry bias)が存在することを示唆している。
コクラン・コラボレーション (Cochrane Collaboration) というのは、治療や予防に関する医療技術を評価する世界各国の研究者が参加しているプロジェクトです。無作為化比較試験を中心に、世界中の臨床試験の結果を収集し、試験の質的評価を行い、統計学的に統合して、その結果を医療関係者や医療政策決定者、さらには消費者に届け、合理的な意思決定に供することを目的としています。
Cochrane Database of Systematic Reviewsと呼ばれるデータベースは、過去の多くの臨床試験などの論文からエビデンスレベルの高いものを集めて吟味する「システマティックレビュー」の手法を使い、その時点での最良の治療法のエビデンスを提示しています。つまり、Cochrane Database of Systematic Reviewsでの結論は、その時点での最も信頼性の高い情報と言えます。
したがって、この論文の「製薬企業が資金提供している臨床試験は、より好ましい有効性の結果および結論を出す傾向にある」という結論は、非常に信憑性の高い情報だと言えます。
承認を受けるための臨床試験のほとんどは製薬企業がスポンサーです。つまり、医薬品承認のための臨床試験の多くは、業界バイアス(industry bias)の存在によって、実際より良い結果を報告している可能性があるという指摘です。
【抗がん剤治療の科学的エビデンスは乏しい】
抗がん剤治療が終わればホスピスケアしかないという標準治療の現状も問題かもしれません。抗がん剤治療とホスピスケアの間を埋める治療手段として代替療法の存在意義も検討する必要があると思います。
代替療法は玉石混淆で、エビデンスの乏しいものも多くありますが、臨床試験で有効性のエビデンスの証明されている治療法も多くあります。
「終末期の抗がん剤治療」や「代用エンドポイントを使った迅速承認で承認された抗がん剤」よりエビデンスの高い代替療法は多数あります。
がん治療専門の腫瘍内科医は、サプリメントや漢方薬や再利用薬を使ったがん治療法はエビデンスが無いと言って拒否しますが、現在使われている抗がん剤の半数以上は、臨床的有用性のエビデンスが無いという事実を理解する必要があります。
「標準治療で認められているがん治療のガイドラインは科学的エビデンスに基づいている」という認識は、必ずしも正しくないと言えます。
新刊紹介

抗がん剤治療が失敗する理由を解説し、その原因に対処する補完医療についても解説しています。さらに、抗がん剤治療の止め時を適切に判断し、終末期における「生活の質」と「死の質」の両方を良くすることの大切さを解説しました。死を早める可能性もある「無駄な抗がん剤治療」を避けるためには、医師の言いなりにならずに、患者自身が正しい知識を得て、もっと考える必要があると思います。
| « 874)抗がん剤... | 876)オピオイ... » |