がんの予防や治療における漢方治療の存在意義を考察しています。がん治療に役立つ情報も紹介しています。
「漢方がん治療」を考える
カレンダー
| 2025年4月 | ||||||||
| 日 | 月 | 火 | 水 | 木 | 金 | 土 | ||
| 1 | 2 | 3 | 4 | 5 | ||||
| 6 | 7 | 8 | 9 | 10 | 11 | 12 | ||
| 13 | 14 | 15 | 16 | 17 | 18 | 19 | ||
| 20 | 21 | 22 | 23 | 24 | 25 | 26 | ||
| 27 | 28 | 29 | 30 | |||||
|
||||||||
goo ブログ
過去の記事
カテゴリ
最新の投稿
最新のコメント
最新のトラックバック
ブックマーク
プロフィール
検索
gooおすすめリンク




| URLをメールで送信する | |
| (for PC & MOBILE) | |
703)延命効果が証明されていない高額な抗がん剤がなぜ使用されるのか
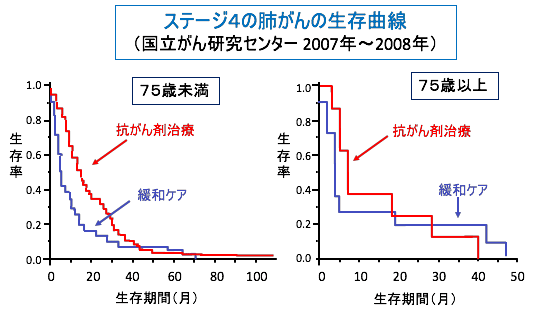
図:ステージ4の肺がんの生存曲線(国立がん研究センター2007年〜2008年)。75歳以上の高齢者では抗がん剤治療が生存率を高めない可能性がある。この解析では、75歳以上では、生存期間が40カ月以上であったのは、抗がん剤治療を受けなかった患者のみであった。(出典:https://www.ncc.go.jp/jp/information/pr_release/2017/0427/index.html)
703)延命効果が証明されていない高額な抗がん剤がなぜ使用されるのか
【1個の抗腫瘍薬を上市するのに3000億円くらいの研究開発費がかかっている】
がん治療薬は開発リスクが高いことが指摘されています。米国のデータでは、2003年から2011年の間に第1相臨床試験の開始がFDA(米国食品医薬品局)に認められた物質のうち、最終的に医薬品として認可されたのは6.7%で、この数値は他の領域の医薬品の半分の成功率と報告されています。
このように開発に失敗する薬が多いので、失敗した分を含めると、一つの抗がん剤を市場に出すまでの研究開発費は平均3000億円以上かかっていると言われています。最近のJAMAの論文に以下のような報告があります。
Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009-2018.(2009年から2018年に新薬を上市するために必要な推定される研究開発投資)JAMA. 2020 Mar 3;323(9):844-853.
2009年から2018年の間に米国食品医薬品局(FDA)によって承認された355種類の新しい治療薬および生物学的薬剤のうち、47社が開発した63種類を解析しています。
その結果、失敗した開発への支出を含めて計算すると、1種類の新薬を市場に出すための研究開発費の推定中央値は9億8500万ドル(95%信頼区間: 6億8360万〜12億2890万ドル)で、推定平均値は13億3590万ドル(95%信頼区間: 10億4250万ドル〜16億3750万ドル)でした。
つまり、途中で失敗した薬の開発費を含めると、1個の新薬を市場に出すための研究開発費の平均は1500億円(13億3590万ドル)くらいという結果です。
5種類以上の薬がある領域別で解析すると、神経系薬剤の推定中央値は7億6590万ドル(95%信頼区間:3億2300万〜14億7350万)から、抗腫瘍性および免疫調節薬の27億7160万ドル(95%信頼区間:20億5180万-53億6620万)の範囲でした。
がん治療の領域では、1種類の抗腫瘍薬を上市するのに2000億円から5500億円くらい(中央値で3000億円くらい)の研究開発費がかかっているという推定です。
動物実験などの基礎研究でがんの治療薬として効果が期待されて臨床試験の許可を得た候補薬のうち、20個に1個程度しか最終的に薬として成功していません。残りは、開発途中で中止になるので、それまでの研究開発費用は無駄になるということです。
米国で2000年から2009年の10年間に認可された新薬は212種類で、そのうちがん治療薬は24種類で、このうち14種類は血液がんの治療薬です。つまり、固形がんの治療薬は特に開発が難しいようです。
がん治療薬の開発は製薬会社にとって非常にリスクが高いので、新薬として認可された薬は、その薬自身の研究開発に費やした費用の何十倍もの研究開発費を回収しないと元が取れないために、がんの新薬は年々高額になっています。
しかし、抗がん剤は製薬メーカーに巨額の利益をもたらしているという実態が報告されています。
【抗腫瘍薬は利益率が高い】
新薬を開発した製薬メーカーは、特許が切れるまで、莫大な利益を得ることができます。抗体薬などの生物学的製剤は特許が切れても、ジェネリックが出にくいので、長期間にわたって利益を得ているようです。以下のような報告があります。
Comparison of Sales Income and Research and Development Costs for FDA-Approved Cancer Drugs Sold by Originator Drug Companies(最初に開発した製薬会社が販売したFDA承認のがん治療薬の売上高と研究開発費の比較)JAMA Netw Open. 2019 Jan 4;2(1):e186875.
【要旨の抜粋】
方法:製薬業界の売上データを使用して、特許またはマーケティング権を保有している会社(オリジネーター企業)の抗がん剤の売上から生じた累積収入を計算した。
1989年から2017年に米国食品医薬品局(FDA)によって承認されたすべての抗がん剤は、米国食品医薬品局のウェブサイトおよび文献から特定された。
項目別の製品販売データは、企業の財務報告から抽出された。
販売データは、インフレ調整後の2017年米ドルで表示された。これらの医薬品の販売による累積収入は、文献から推定された研究開発費と比較された。
結果:米国食品医薬品局(FDA)が承認した156種類のがん治療薬のうち、99種類の薬(63.5%)が承認から半年以上経過しており、今回の分析に含まれた。販売期間の中央値は10年(範囲、1〜28年)であった。
文献で推定されている医薬品1個あたりのリスク調整後の研究開発費用の中央値は7億7,400万ドル(範囲、28億2700万-2億1900万ドル)であるのに対し、2017年末までに、累積売上高の中央値は研究開発投資額1ドルあたり14.50ドル(範囲、3.30ドル-55.10ドル)であった。
リスク調整後の研究開発コストを回収するまでの時間の中央値は5年(2〜10年、n = 56)であった。
がん治療薬は、特に生物学的製剤においては、特許が切れて市場独占が終了した後においても、開発製薬企業に10億ドル規模の利益を生み出し続けた。
結論:抗がん剤は高価格で、開発企業において、研究開発コストをはるかに超える利益を生み出している。
1989年から2017年までにFDAによって承認された99種類の抗がん剤に関する研究です。これらの抗がん剤の約3分の1は、1年間の販売額が10億ドルを超えています。2017年末までの収入の中央値は、研究開発費1ドルごとに14.50ドル(範囲、3.30〜55.10ドル)でした。
Originator Drug Companiesというのは、新薬を開発し、特許を持っている製薬メーカーで、特許が切れるまで独占して販売できるため、巨額な利益を得ることができます。
新薬の物質特許が切れた後、後発医薬品メーカーは、新薬と同じ有効成分で効能・効果、用法・用量が同一で新薬に比べて低価格な医薬品、いわゆる「ジェネリック医薬品」を発売しますので、先発メーカーの利益は減少します。しかし、生物学的製剤のように、製造法が特殊な医薬品などは、ジェネリック医薬品ができにくく、特許および独占販売権の満了後も、薬を最初に開発した企業に高利益を生み出し続けます。
製薬産業は最も利益率が高い産業領域と考えられています。
昔から「薬9層倍(くすりくそうばい)」と言う言葉があります。「薬の売値は原価の九倍もする」という意味で、儲もうけが大きいこと、暴利をむさぼるたとえで使われます。
【研究開発費よりマーケティングの費用の方が多い】
製薬会社は販売促進の費用をできるだけ少なく公表しようとしており、それは薬の販売促進に莫大な費用を使っていることが明らかになると非難されるからです。つまり、薬の販売促進に多額の費用を使っていることに後ろめたさがあるからです。
しかし実際は、販売促進のための費用が研究開発の費用より多いことが報告されています。
以下のような論文があります。
The Cost of Pushing Pills: A New Estimate of Pharmaceutical Promotion Expenditures in the United States(医薬品の販売促進のコスト:米国の医薬品販売促進費の新しい見積り)PLoS Med. 2008 Jan; 5(1): e1.
米国では、販売促進を目的にした説明会や講演会の数は1998年の120,000回から2004年の371,000回に増え、2000年には314,000回の販売促進のための講演会や会議で19億ドルが使われています。
この論文では、2004年の製薬企業の販売促進のための費用を推定しています。
米国国内売上高の2,354億ドルのうち、売上高の24.4%を販売促進、13.4%が研究開発で消費していると推定しています。他にもいくつか報告があり、売上げの20から30%くらいを販売促進に使っており、研究開発費よりも多いというのが結論です。販売促進費が売上高の30%以上という報告もあります。
この論文の結論は、製薬企業が販売促進に費やす費用は、研究開発費の2倍くらいと言っています。
製薬企業は研究開発費に多くを使っていると主張していますが、実際は販売促進の費用の方が多いのが事実のようです。
【抗がん剤は生存期間を長くすることを目標にすると、毒性が強くなる傾向がある】
生存期間の延長の引き換えに毒性(副作用)が強くなることは抗がん剤治療においては多く経験されます。以下のような報告があります。
The Price We Pay for Progress: A Meta-Analysis of Harms of Newly Approved Anticancer Drugs(進歩の対価:新たに承認された抗がん剤の有害性のメタ分析)J Clin Oncol. 2012 Aug 20;30(24):3012-9
【要旨の抜粋】
目的:新しい抗がん剤の承認は、ランダム化比較試験によって、それまでの標準治療と比較して有効性が向上している結果を示すことが必要である。この際、毒性(副作用)に関してはあまり重視されていない。新たに承認された抗がん剤の毒性を分析する。
患者と方法:2000年から2010年の間に米国食品医薬品局(FDA)によって承認された固形腫瘍の治療薬を評価するランダム化比較試験を特定した。安全性と忍容性の3つのエンドポイント(治療に関連する死亡、毒性に関連する治療中止、グレード3または4の有害事象)について検討した。
結果:38 件のランダム化試験が分析された。対照群と比較して、毒性による死亡のオッズ比(OR)は新しい薬剤の方が高かった(OR、1.40; 95%CI、1.15〜1.70; P <.001)。毒性による治療中止のオッズ比も同様に新しい薬剤の方が高かった(OR、1.33; 95%CI、 1.22〜1.45、P <.001)。
グレード3または4の有害事象のオッズ比(OR、1.52; 95%CI、1.35〜1. 71; P <.001)も、新薬の方が高く、特に下痢、皮膚反応、神経障害などの非血液学的有害事象で発現が高かった。安全性エンドポイントと全生存期間または無進行生存期間の間に有意な相関関係は認めなかった。
結論:全生存期間や無増悪生存期間というエンドポイントの改善につながる新しい抗がん剤は、有害事象(副作用)の発生率と治療関連の死亡率も増加させる。
新薬の多くは、全生存期間や無増悪生存期間は延長しても、有害事象(副作用)の発生率と治療関連の死亡率も増加させているという論文です。
つまり、抗がん剤の新薬は、延命効果と引き換えに、副作用による苦しみも増えているということです。
【メリットの少ない高額な抗がん剤が使われている】
米国国立がん研究所の研究者の論文によると2002年から2014年の間に承認された72のがん治療法は、それ以前の古いがん治療薬と比較して、2.1ヶ月の延命効果を示したにすぎないことを示しています。(JAMA Otolaryngol Head Neck Surg. 2014;140(12):1225-1236. )
2008年から2012年の5年間に代用エンドポイント(奏功率や無増悪生存など)の結果でFDAから承認された36種類の抗がん剤のうち、市販後調査で18種類は全生存期間を延ばす有効性が認められていません。(JAMA Intern Med. 2015 Dec;175(12):1992-4.)
この18種類の抗がん剤の関しては、生活の質を改善したものも少なく、中にはQOLを悪化したものもありました。全生存期間に関しては、生存期間を明らかに短縮したものもありました。
つまり、代用エンドポイントで承認された抗がん剤の半分は全生存期間を延ばす効果は認められず、QOLを悪化したり、生存期間を短縮したものもあったという結果です。
延命効果もなく、QOLの改善効果もなく(QOLを悪化したものもある)承認されている6種類の抗がん剤の平均の費用は、1年間で8万7922ドル ($20, 237 〜 $169, 836)でした。
つまり、現在使用されている抗がん剤の中には、生存やQOLに全く利益が無いのに、1年間に1000万円以上の費用をかけて使われているものがあるということです。(JAMA Intern Med. 2017;177(2):276-277.)
米国臨床腫瘍学会は、少なくとも2.5か月間、寿命を延ばすか腫瘍を制御するという新しい抗がん剤の目標を設定しました。しかし、2014年から2016年に承認された抗がん剤のうち5分の1しかこの基準を満たしていませんでした。以下のような報告があります。
An Appraisal of Clinically Meaningful Outcomes Guidelines for Oncology Clinical Trials.(腫瘍学臨床試験のための臨床的に意義のある結果ガイドラインの評価)JAMA Oncol. 2016;2(9):1238-1240.
米国臨床腫瘍協会(ASCO)が、膵臓がん、肺がん、大腸がん、乳がんの臨床試験に有意義な目標を設定しました。この「臨床的に意味のある改善」というのは、それまでの標準治療に比較して、生存期間が25%以上かつ絶対値で2.5ヶ月以上の延長というものです。
それまでの標準治療(対照群)の生存期間が20ヶ月であれば、25%の延長で5ヶ月間以上の延命効果が認められれば、「臨床的に意味のある改善」と評価するということです。この程度の効果があれば、それまでの治療薬と比べて、意味のある有効性がある新薬という評価です。
それまでの標準治療(対照群)の生存期間の平均が8ヶ月であれば25%では2ヶ月ですが、この場合は絶対値の2.5ヶ月が必要です。
より野心的な目標は非現実的で達成不可能と考えられたため、この選択された目標は意図的にかなり控えめです。今までの治療薬に比較して50%以上の生存期間の延長を達成する新薬の開発など、極めて困難と認めているようなものです。
さらに重要なことは、これらの目標は国の承認や保険の適用を意図したものではありません。患者と医師が臨床試験の結果に満足する目標値です。
したがって、ASCOが目標とする「臨床的に意味のある改善」のレベルに達していなくても、非治療群を対照にした比較で統計的有意差が認められれば、たとえ2週間の延命でも薬として承認されます。
2014年4月1日から2016年2月29日までの間に、米国食品医薬品局(FDA)はがんの治療のために47の治療法を承認しました。 この47種類の治療薬のうち、「全生存期間の25%かつ2.5ヶ月以上の延長」という「臨床的に意味のある改善」という基準を満たしたものは9つの治療(19%)のみでした。
つまり、それまでの標準治療に比べて25%以上の生存期間の延長を達成できたのは新たに承認された抗がん剤の19%に過ぎないという結果です。
【高齢者の抗がん剤治療は死を早める可能性もある】
国立がん研究センターと厚労省、経済産業省が主体となり調査を実施しています。平成19年から20年に同センター中央病院を受診したがん患者約7000人のうち、70歳以上の高齢者約1500人が対象です。
がんの種類別に、抗がん剤による治療を中心に行った場合と、痛みを和らげる「緩和ケア」に重点を置いた場合とで、受診から死亡までの期間(生存期間)を比較しました。
その結果、主に肺がん・大腸がん・乳がんで末期(ステージ4)の高齢患者の場合、抗がん剤治療の有無にかかわらず、生存率は同程度であり、抗がん剤治療が明確な効果を示さない可能性があると言っています。
例えば肺がんの場合、生存期間が40カ月以上のグループは抗がん剤治療を受けなかった患者のみでした。同様に75歳以上で見た場合、10カ月以上生存した人の割合は、抗がん剤治療を受けなかった患者の方が高く、生存期間も長いという結果でした。(トップの図)
「抗がん剤治療が明確な効果を示さない可能性がある」と控えめな表現になっていますが、「肺がんの場合、70歳以上では生存期間が40カ月以上のグループは抗がん剤治療を受けなかった患者のみだった。75歳以上では、10カ月以上生存した人の割合は、抗がん剤治療を受けなかった患者の方が高く、生存期間も長かった。」という結果は、「70歳以上の肺がんでは、抗がん剤治療を受けると早く死ぬ」と言っているのと同じです。
将来的に画期的な薬ができて、75歳以上でも副作用が少なく、有効性が高い治療ができれば、75歳以上でも抗がん剤治療を受けるメリットは出てきます。 問題は、現時点の抗がん剤治療は75歳以上には「メリットがない」、あるいは「死を早めている可能性がある」ということです。
現行の抗がん剤治療は、体力と回復力に依存した治療法と言えます。体力と回復力が高い若い人の場合は、副作用に耐えられる分、延命効果は得られます。副作用で苦しんでも少しでも長く生きたければ、若い人は現行の最大耐用量の抗がん剤治療は意味があります。
しかし、体力も回復力も低下している高齢者の場合は、副作用によるデメリットが、抗腫瘍効果によるメリットより大きいので、抗がん剤治療は意味が無いということです。
【米国食品医薬品局(FDA)承認の抗がん剤の半数以上は臨床的有益性がない】
米国では医薬品の承認は食品医薬品局(FDA)が行っています。
FDAによる新薬承認のハードルは日本に比べてかなり高く、臨床的なエビデンス(証拠)が十分になければ承認されないと一般的には考えられています。しかし、抗がん剤の場合は事情が異なるようです。
フランスと米国の疫学や統計学が専門の研究グループが、2000年から2015年の間にFDAによって承認された進行した固形がんを治療するための新しい抗がん剤および生物製剤の全てについて、臨床的有用性を総合評価しています。結論は、FADが承認している抗がん剤の多くが十分な臨床的有益性が認められないというものでした。
以下のような報告があります。
Clinical benefit, price and approval characteristics of FDA-approved new drugs for treating advanced solid cancer, 2000-2015. (進行した固形がん治療のためのFDA承認の新薬の臨床的利益、価格、承認特性、2000-2015)Ann Oncol. 2017 May 1;28(5):1111-1116.
【要旨の抜粋】
材料と方法:2000年から2015年の間にFDAによって承認された進行した固形がんを治療するための新しい抗がん剤および生物製剤をすべて含めて検討した。
医薬品の臨床的利点は、米国臨床腫瘍学会の臨床価値枠組み(ASCO-VF )の2016年更新版および欧州腫瘍学会の臨床利益スケール(ESMO-MCBS)を用いて、主要な臨床試験のFDA医学的レビューに基づいてグレード化した。
薬物の特性および承認は、公的に入手可能なFDA文書から得られ、価格は、米国メディケア、米国退役軍人健康管理および英国市場システムによって評価された。
結果: 2000年から2015年までの間に、FDAは固形がんの治療のための51個の新薬を承認した。 そのうち37個の薬物(73%)の価値を評価することができた。ESMO-MCBSの指標では、5個(14%)がグレード1(最低)、9個(24%)がグレード2、10個(27%)がグレード3、11個(30%)がグレード4、2個(5%)がグレード 5(最高)と評価された。したがって、13の薬物(35%)は有意な臨床的有益性を示した(スケールレベル4および5)。
ASCO-VF(3.4-67の範囲を有する)による評価では、薬物評価の中央値は37(20-52)であった。臨床的利益と薬価との間には相関が見られなかった(P = 0.9)。 薬物および承認の特徴は、臨床的利益と有意に関連していなかった。
結論: FDAが最近認可した新しい抗がん剤の多くは、現在の評価スケールで測定されたように、高い臨床的利益をもたらさなかった。 薬物価格と社会および患者の利益との間には関係は認めなかった。
抗がん剤の有効性を評価する指標として、「生存期間の延長」と「生活の質(QOL)の改善」の両方が重要です。ある抗がん剤治療で生存期間が3ヶ月延長しても、副作用が強く、苦しんでの3ヶ月延長(苦しむ期間は抗がん剤治療期間全体なのでもっと長い)では、臨床的に意味のある有益性があるとは評価できません。副作用が少なく、1年間の生存期間の延長が得られる治療であれば、臨床的有益性はかなり高いと評価できます。
費用も評価の対象になります。平均生存期間が6ヶ月から8ヶ月に2ヶ月延長するときに8ヶ月間の薬剤費が2000万円だとしたら、私は受ける気がしません。
日本の場合は、国民皆保険制度と高額療養費制度などによって、1ヶ月に何百万円もする抗がん剤治療でも自己負担は数万円ですむので、気兼ねなく高額な新薬が使用されています。
このように、抗がん剤の有益性の評価は、生存期間の延長の程度や副作用やQOLの改善度や費用などを総合的に検討する必要があります。
この目的で、米国臨床腫瘍学会(ASCO)や欧州腫瘍学会(ESMO)は、がん治療の臨床的利益を定量化するための枠組み(Value Framework;VF)や臨床利益スケール(Magnitude of Clinical Benefit Scale;MCBS)を作成しています。
この論文では、米国臨床腫瘍学会の臨床価値枠組み(ASCO-VF)の2016年更新版および欧州腫瘍学会の臨床利益スケール(ESMO-MCBS)で、FDAが2000年から2015年の間に承認した抗がん剤を臨床的有益性の観点から分析しています。
SCO-VFとESMO-MCBSはいずれも生存率改善や毒性やQOLを定量化し、ASCO-VFでは130点満点のスコアで、ESMO-MCBSでは5段階のグレードで評価しています。
その結果、ESMO-MCBSではグレード評価では、評価できた37個の抗がん剤のうち有意な臨床的有益性(スケールレベル4および5)が認められたのは13個(35%)で残りの65%の薬は有意な臨床的有益性があるとは評価されないという結果でした。
また、臨床的利益と薬価との間には相関が見られないことも明らかになっています。高い薬が効くわけでも無いということで、臨床的に有益でないのに極めて高価な薬剤が承認されていることを示しています。
【多くの抗がん剤が延命効果を証明せずに承認されている】
延命効果も生活の質の改善効果も認められない抗がん剤を国が認める訳が無いと思うかもしれません。しかし、あるトリックによって、全く有益性の無い抗がん剤が認可されて、標準治療で使用されていることが明らかになっています。
そのトリックというのは、生存期間の延長でなく、奏功率や無増悪(あるいは無再発)生存期間といった代用エンドポイント(surrogate endpoint)で効果を示して承認されているからです。
全生存期間の延長を証明するには時間がかかるので、有効な治療法が少ないがん治療の臨床現場に新薬を早く届けるために、全生存期間の延長を予想できる奏功率や無増悪生存期間で代用しています。
「腫瘍が縮小すれば生存期間も延びるだろう」「再発や増悪するまでの期間が延長すれば生存期間も延びるだろう」という予測に基づいています。
しかし、奏功率および無増悪生存期間と全生存期間との相関は低いことが明らかになっています。つまり、奏功率(がん組織の縮小率)が高くても延命効果が無いことが多く、無増悪生存期間は延びても全生存期間は延びないことは多くあると言うことです。
腫瘍が一時的に縮小して、増悪しない期間が延長しても、抗がん剤の毒性による副作用によって、最終的には全生存期間は同じくらいという結果になっているということです。抗がん剤を使うと、一時的にはがん細胞の増殖は抑えられますが、薬剤耐性の悪性度の高いがん細胞が残るので、増殖しだすと抗がん剤を使わなかった場合より増殖が早いという理由もあります。
そして、FDAは製薬会社に承認後に生存期間の延長を証明する臨床試験を行うことを義務づけていないという問題点も指摘されています。
代用エンドポイントを根拠に承認されたがん治療薬とその後の全生存期間に関して、FDA(米国食品医薬品局)の5年間の承認薬を解析した論文があります。
Cancer Drugs Approved on the Basis of a Surrogate End Point and Subsequent Overall Survival: An Analysis of 5 Years of US Food and Drug Administration Approvals(代用エンドポイントを根拠に承認されたがん治療薬とその後の全生存期間:米国食品医薬品局の5年間の承認薬の解析) JAMA Intern Med. 2015; 175(12): 1992-1994.
【論文内容の抜粋】
抗がん剤の最近の承認の多くは、奏効率や無増悪生存期間などの代用エンドポイントに基づいて行われている。承認が代用エンドポイントに基づいている場合は、その後の臨床試験によって全生存期間の延長を確認することが望まれており、それは義務でもある。
どれくらいの数のがん治療薬が代用エンドポイントに基づいて承認されているか、これらの薬のその後の試験が報告されているか、それらの薬物が全生存期間を改善しているかを調べた。
方法:2008年1月1日から2012年12月31日まで、FDAによるすべての承認薬を検討した。承認された経路(迅速承認と通常承認)および代用エンドポイント(奏功率または無増悪生存期間)に使用について特定した。
代用エンドポイントに基づいて承認されたすべての医薬品について、2015年8月22日の時点で公開された文献を体系的に検索し、全生存期間に関する効果の報告を特定した。
結果:検索期間内に54個のがん治療薬の承認が確認され、代用エンドポイントに基づいて承認されたのは36個の薬剤(67%)であった。15の迅速承認では全てが代用エンドポイントに基づき、通常の承認では、39個中21個(54%)が代用エンドポイントに基づいて承認されていた。
代用エンドポイントに基づいて承認された36個の薬剤のうち、19個(53%)は奏功率(腫瘍体積の縮小)が主要エンドポイントとして使用され、36個中17個(47%)は無増悪生存期間あるいは無再発生存期間が主要エンドポイントとして使用されていた。
中央値4.4年の追跡期間で、5個の薬(迅速承認15個中1個、通常の承認21個中4個)がその後のランダム化臨床試験で全生存期間を改善することが示された。18個の薬剤(迅速承認15個中6個、通常承認21個中12個)は、全生存期間の延長を示すことができず、13の薬物は全生存期間に対する効果が不明であった。
考察:調査期間の間に、承認された抗がん剤54個のうち、代用エンドポイントに基づいて承認されたのは36個(67%)であった。数年間の追跡調査で、代用エンドポイントに基づいて承認された36個のうち31個(86%)(承認された54の薬剤の57%)は生存期間を延長する効果がなかったか、不明であった。
これらの結果は、承認された抗がん剤の多くが生存期間の延長を証明できていないことを示している。
2008年以来、FDAはこれまで以上に多くの薬剤を承認しており、抗がん剤は代用エンドポイントに基づいて承認しているが、この代用エンドポイントは全生存期間との相関が低い。
我々の結果は、FDAが生存期間を延長しない高価で毒性の高い多くの薬物を承認している可能性があることを示唆している。 したがって、市販後の臨床試験の実施は非常に重要である。
米国では抗がん剤治療の費用の平均は1ヶ月あたり約1万ドル(約112万円)と言われており、抗がん剤治療による延命期間の平均は42日と言われています。
この10年間にFDAは50以上の抗がん剤を承認していますが、臨床試験で全生存期間の延長を証明して承認された薬は一部で、3分の2以上のがん治療薬は奏功率(腫瘍の縮小率)や無増悪生存期間といった代用エンドポイントで効果を示して承認されています。そして、承認後の臨床試験で有効性が証明できずに承認が取り消された抗がん剤も多くあります。
一つの例として、転移性乳がん患者の無増悪生存期間に基づいて迅速承認を受けたベバシズマブ(商品名アバスチン)があります。その後の試験では、全生存期間の延長は認められず、重大な毒性が認められたため、承認が取り消されています。(日本では使用されています)
2009年の連邦政府監査院の報告書は、代用エンドポイントで承認された医薬品の市販後調査の実施を強制していないことに対して米国食品医薬品局(FDA)を批判しています。
この論文の著者も、FDAが生存期間を延長しない高価で毒性の高い多くの薬物を承認している可能性があると非難しています。FDAは製薬会社に承認後に生存期間の延長を証明する臨床試験を行うことを義務づけていないことが問題だと言っています。
米国食品医薬品局(FDA)が承認した薬は間違いないというFDAへの信頼は抗がん剤に関しては間違いのようです。
【欧州医薬品庁(EMA)が承認した抗がん剤の半分以上は有益性を示していない】
「新しいがん治療薬の半分以上は生存と健康に何の利益も示さない」というのは米国だけでなく、欧州でも問題になっています。
2009年から2013年に承認された48のがん治療薬のうち、半数以上はほとんど有益性が認められず、有益性が認められた場合でもそれは臨床的に意味の無いレベルであることが明らかになっています。以下のような報告があります。
Availability of evidence of benefits on overall survival and quality of life of cancer drugs approved by European Medicines Agency: retrospective cohort study of drug approvals 2009-13(欧州医薬品庁によって承認された抗がん剤の全生存期間および生活の質に関する有効性の証拠の入手可能性:2009年から20013年の薬物承認の後ろ向きコホート研究)BMJ. 2017; 359: j4530.
【要旨の抜粋】
目的:欧州で承認されたがん治療薬の全生存期間および生活の質(QOL)に関するデータの入手可能性を明らかにする。
設定:2009年から2013年までの欧州医薬品庁(EMA)によるがん治療薬の承認に関する公的に入手可能な規制および科学的報告書。
主なアウトカム指標:試験デザイン(無作為化、クロスオーバー、盲検化)、対照薬、エンドポイントに応じたがん治療薬の中心的試験および市販後臨床試験。 承認時および市販後に評価された全生存または生活の質に対する有効性の利用可能性および規模。 がん治療薬の報告された利益の臨床的価値を評価するために欧州臨床腫瘍学会の臨床効果尺度(ESMO-MCBS)を使用。
結果:2009年から2013年の期間に欧州医薬品庁(EMA)が承認したがん治療薬は48製剤、68適応であった。このうち8適応(12%)は、治験薬だけの単群試験に基づいて承認されていた。
承認時点で生存期間の有意な延長が認められたのは、24/68適応(35%)で、その延長期間は1.0~5.8ヵ月(中央値2.7ヵ月)であった。また、承認時点でQOLの改善が認められたのは、7/68適応(10%)であった。
承認時点で全生存期間の延長の証拠がなかった44適応のうち、市販後の臨床試験で全生存期間延長のエビデンスが確認されたのは3適応(7%)、QOLに対する有益性が報告されたのは5適応(11%)であった。
また、欧州医薬品庁が承認した68適応について、承認後中央値で5.4年(3.3~8.1年)追跡した結果、全生存期間または生活の質(QOL)の有意な改善が示されたのは35適応(51%)に過ぎず、残りの33適応(49%)は不明なままであった。
また、生存期間に関して有益性が認められた23適応のうち、ESMO-MCBSスケールで臨床的に意味があると判定されたのは半数未満(11/23適応、48%)であった。
結論:2009〜13年の欧州医薬品庁(EMA)によって承認されたがん治療薬の体系的評価は、ほとんどの医薬品が生存または生活の質に関する利益の証拠なしに市場に参入したことを示している。市販後最低でも3.3年後に、これらの薬剤ががん患者の生存を延長したり生活の質を改善したという決定的な証拠はまだ無い。既存の治療法やプラセボに比べて生存期間の延長がみられた場合でも、それらは多くの場合極めて限定的であった。
2009年から2013年の期間に欧州医薬品庁(EMA)が承認したがん治療薬は48製剤、68適応でした。この「適応」というのは、この論文の「indications」の日本語訳です。使用が承認された病気や症状を「適応」や「適応症」いいます。ある抗がん剤が胃がんと肺がんで承認されれば1つの薬剤で2つの適応になります。
承認時点で生存期間の有意な延長が認められたのは、24/68適応(35%)で、その延長期間は1.0~5.8ヵ月(中央値2.7ヵ月)でした。また、承認時点でQOLの改善が認められたのは、7/68適応(10%)でした。
承認時点で全生存期間の延長の証拠がなかった44適応のうち、市販後の臨床試験で全生存期間延長のエビデンスが確認されたのは3適応(7%)、QOLに対する有益性が報告されたのは5適応(11%)でした。
また、欧州医薬品庁が承認した68適応について、承認後中央値で5.4年(3.3~8.1年)追跡した結果、全生存期間または生活の質(QOL)の有意な改善が示されたのは35適応(51%)に過ぎず、残りの33適応(49%)は不明なままでした。
また、生存期間に関して有益性が認められた23適応のうち、欧州腫瘍学会の臨床利益スケール(ESMO-MCBS)で臨床的に意味があると判定されたのは半数未満(11/23適応、48%)でした。この論文の結果は以下の図にまとめています。
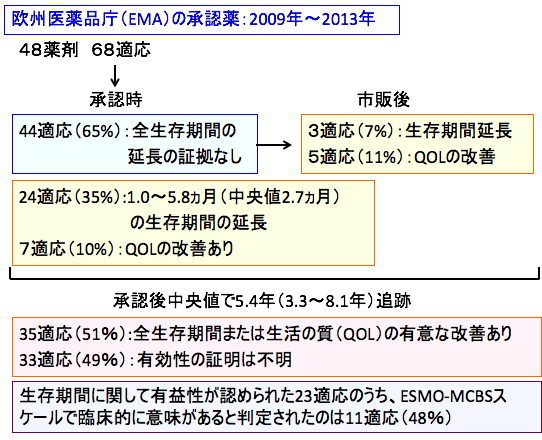
図:2009年から2013年の期間に欧州医薬品庁が承認したがん治療薬は48製剤、68適応であった。承認時点で生存期間の有意な延長が認められたのは24/68適応(35%)で、その延長期間は1.0~5.8ヵ月(中央値2.7ヵ月)であった。承認時点でQOLの改善が認められたのは7/68適応(10%)であった。承認時点で全生存期間の延長の証拠がなかった44適応のうち、市販後の臨床試験で全生存期間延長が確認されたのは3適応(7%)、QOLに対する有益性が報告されたのは5適応(11%)であった。承認後中央値で5.4年(3.3~8.1年)追跡した結果、全生存期間または生活の質(QOL)の有意な改善が示されたのは35適応(51%)で、残りの33適応(49%)は有効性が不明なままであった。生存期間に関して有益性が認められた23適応のうち、欧州腫瘍学会の臨床利益スケール(ESMO-MCBS)で臨床的に意味がある有益性があると判定されたのは11/23適応(48%)であった。つまり、承認された68適応のうち臨床的に意味のある生存期間の延長は11適応(16%)しか無かった。(出典:BMJ. 2017; 359: j4530.)
つまり、2009〜13年の欧州医薬品庁(EMA)によって承認されたがん治療薬は、ほとんどの医薬品が生存または生活の質に関する利益の証拠なしに市場に参入したことを示しています。市販後にこれらの薬剤ががん患者の生存を延長したり生活の質を改善したという証拠が得られた薬は少数で、既存の治療法やプラセボに比べて生存期間の延長がみられた場合でも、それらは多くの場合極めてわずかな有益性でした。
この論文の結論の最後の文章は『臨床的に意味のある有益性がない高価な医薬品が承認され、公的資金で運用されている医療システムで支払われると、個々の患者に害が生じ、重要な社会資源が無駄になり、正当で適切な費用の医療の提供が損なわれる』と記述されています。
【統計的に有意差があっても、臨床的に意味のある有益性の無い抗がん剤が多い】
生存期間の延長で統計的に有意差を示しても、臨床的に意味のある有益性があるとは限りません。
生存期間が12ヶ月から14ヶ月に延長しても、強い副作用を伴い、生活の質が著しく低下するような治療薬であれば、臨床的に意味がある有益性があるとは言えないと思います。
固形腫瘍に使用されている71の薬物による生存期間の延長の中央値はわずか2.1ヶ月という報告もあります。(JAMA Otolaryngol Head Neck Surg 2014;140:1225–36.)
多くのがん治療薬が迅速に承認されていますが、患者の生存を改善する十分な証拠を得て市場に参入する抗がん剤はほとんど無いのが実情のようです。
欧州臨床腫瘍学会(ESMO)はがん治療法を評価するツールとして臨床的ベネフィット・スケール・マグニチュード(Magnitude of Clinical Benefit Scale:MCBS)を発表しています。
ESMO-MCBSは、がん治療薬の治療効果を評価するために設計されており、有効性(全生存期間と無増悪生存期間の絶対的な増加およびハザード比の95%信頼区間の下限)と、生活の質(QOL)または毒性をそれぞれ検討します。新規治療法のデータは、病状ごとの予後に関して分析されて臨床的利益が評価されます。
例えば、根治(cure)を目指す治療(手術前の抗がん剤治療や手術後の補助化学療法)では、3年以上の追跡で何%の生存率の増加があるかで有効性が評価できます。
進行がんの緩和的化学療法では、対照群が12ヶ月以下の生存期間の場合、3ヶ月以上の生存期間の延長があれば臨床的に意味がある有用性があると言えます。対照群が12ヶ月以上の生存期間の場合、臨床的に意味があるというには5ヶ月以上の延命が必要かもしれません。
このように、病気の進行状況に応じて、どの程度の生存期間の延長や生活の質の改善や副作用(毒性)を評価して、臨床的に意味のある有用性を評価するツールがESMO-MCBSです。
ESMO-MCBSの評価法を使って、最近承認われた抗がん剤やランダム化比較試験を検証すると、臨床的に意味のある有用性を示した抗がん剤は2割以下のようです。
例えば、以下のような報告があります。
Do Contemporary Randomized Controlled Trials Meet ESMO Thresholds for Meaningful Clinical Benefit?(最近の無作為化比較試験は意味のある臨床的利益のためのESMO閾値を満たしているのか?)Ann Oncol. 2017 Jan 1;28(1):157-162.
【要旨の抜粋】
背景:欧州臨床腫瘍学会(ESMO)は、固形腫瘍に対する化学療法の有効性を評価するツールとして臨床的ベネフィット・スケール・マグニチュード(ESMO Magnitude of Clinical Benefit Scale:ESMO-MCBS)を最近発表した。この研究では、最近報告されているランダム化比較試験がESMO-MCBSで評価される「意味のある臨床的有益性」の閾値に達しているかどうかを評価した。
方法:2011年から2015年の間に論文に公表された乳がん、非小細胞性肺がん、結腸直腸がん、膵臓がんに対する化学療法の有効性を検討したランダム化比較試験を解析した。臨床試験の特徴と結果に関するデータを抽出し、これらのデータをESMO-MCBSで評価した。
結果:対象となるランダム化比較試験は277件(乳がん40%、非小細胞性肺がん 31%、結腸直腸がん22%、膵臓がん6%)であった。サンプルサイズ(対象になった人数)の中央値は532で、83%は製薬企業からの資金提供を受けていた。
277件のランダム化比較試験の中で、138件(50%)の試験で治療群は対照群より統計的に優位であった。これら有効の結果が得られた試験のわずか31%(43/138)の結果がESMO-MCBSによる臨床的に意味のある利益閾値を満たした。
治癒的意図を有する治療のランダム化比較試験では、有効性を示した31件中19件(61%)で意味のある臨床的有益性の閾値を満たしていた。一方、緩和目的の抗がん剤治療のランダム化比較試験では、有効性を示した107件中24件(22%)が臨床的に意味のある有益性の閾値を満たしていた。
ESMO-MCBSが適用され得る226件のランダム化比較試験のうち、ESMO-MCBSの有益性の閾値を満たすことができる臨床試験のデザインで臨床試験を行っていたのは31%(70/226)であった。
結論:統計的に有意な有効性を示した最近のランダム化比較試験のうち、欧州臨床腫瘍学会による臨床的ベネフィット・スケール・マグニチュード(ESMO-MCBS)の基準で臨床的に意味のある有益性の閾値に達したのは3分の1以下であった。これは全ての公開された試験の15%に過ぎない。
研究者や資金提供機関や規制機関や製薬業界は、今後のランダム化臨床試験の設計において、意味のある臨床的利益のために、より厳しい基準を採用すべきである。
この論文の結果は下図にまとめています。

図:(左)欧州臨床腫瘍学会(ESMO)はがん治療法を評価するツールとして臨床的ベネフィット・スケール・マグニチュード(MCBS)を発表している。ESMO-MCBSは、がん治療薬の治療効果を評価するために設計されており、全生存期間と無増悪生存期間の延長、生活の質または毒性をそれぞれ検討し、病状ごとの予後(対照群での治療奏効期間または生存期間)に関して分析され、臨床的有益性を総合評価する。コストは考慮されない。
(右)2011年から2015年の間に論文に公表された乳がん、非小細胞性肺がん、結腸直腸がん、膵臓がんに対する化学療法の有効性を検討したランダム化比較試験は277件で、このうち138件(50%)の試験で治療群は対照群より統計的有意な有効性を示した。これら有効な結果が得られた試験138件中でESMO-MCBSによる臨床的に意味のある利益閾値を満たしたのは43件であった。これは公開された全ての試験の15.5%に過ぎない。(出典:Ann Oncol. 2017 Jan 1;28(1):157-162.)
別の研究グループからも同様の調査結果が報告されています。2011から2016年に欧州医薬品庁からに承認を受けた38種類のがん治療薬に対する70件の臨床試験を、欧州臨床腫瘍学会の臨床利益スケール(ESMO-MCBS)で評価しています。
Five years of EMA-approved systemic cancer therapies for solid tumours-a comparison of two thresholds for meaningful clinical benefit. (固形腫瘍のための欧州医薬品庁承認の全身がん治療法の5年間 – 意味のある臨床的利益のための2つの閾値の比較。)Eur J Cancer. 2017 Sep;82:66-71.
ESMO-MCBSは、がん治療薬の治療効果のレベルを評価するために設計されています。最初に発表されたESMO-MCBSとその改良版によって定義された「意味のある臨床的利益」の閾値を満たすものがどの程度存在するかを検討しました。
その結果、「意味のある臨床的利益」があると評価されたがん治療薬は、最初のESMO-MCBSの基準では21%、改良版の基準では11%しかありませんでした。つまり、基準の違いによって評価は変わりますが、承認されたがん治療薬の80〜90%は臨床的に意味のある有益性を示さないという結果です。
承認された薬の8割以上が、なぜ臨床的に意味のあるメリットを示す証拠を提示していないのに承認されたのか理解に苦しむのですが、これが事実なのです。
【抗がん剤の多くは生存期間や生活の質の向上に役立っていない】
代用エンドポイントで承認された抗がん剤のうち、市販後に全生存期間の延長や生活の質の改善が証明できたのは極めて少数です。その結果、がん患者が実際に投与されている抗がん剤の多くは生存期間や生活の質の向上に役立っていない可能性があるという事実が明らかになっています。
米国食品医薬品局(FDA)や欧州医薬品庁(EMA)が承認したがん治療薬のうち、約3分の2は生存率の向上または生活の質の改善の証拠が承認時に示されていませんでした。十分な証拠がなく承認された抗がん剤のうち、市販後の臨床試験で既存の薬やプラセボと比較して生存率の改善が認められたものは20%以下です。つまり、現在使用されている抗がん剤の半数以上は全生存期間や生活の質の改善にメリットが無いことを示しています。
かりに薬物が生存期間を延長しても、その利益はしばしば僅かです。固形腫瘍に使用されている71の薬物による生存期間の延長の中央値はわずか2.1ヶ月でした(JAMA Otolaryngol Head Neck Surg 2014;140:1225–36.)。
承認済みの抗がん剤の多くは、信頼性が不十分な代用エンドポイントに基づいており、市販後の研究では、患者中心のエンドポイントでこれらの薬剤の有効性および安全性が確認されることはめったにありません。
これに加えて、がん治療薬の費用の平均は年間1000万円を超えています。これは、確実な毒性を有し、利益が不確実ながん治療薬に対して莫大な支出を行っていることを意味します。
【無増悪生存期間と全生存期間の相関は低い】
新薬の承認を得るためには、臨床試験で有効性と安全性を証明する必要があります。有効性の証明において、抗がん剤の場合は、生存期間の延長か生活の質(QOL)の改善が必要です。
しかし、生存期間の延長を確認するためには時間がかかるので、新薬の承認を迅速化するために、「生存期間の延長」を推測できる代用エンドポイントを使って評価し、この代用エンドポイントで有効性が証明されれば承認するということが行われています。
臨床試験によって治療の有効性を評価する項目をエンドポイント(endpoint)と言います。
抗がん剤の臨床試験で本来求めたいアウトカム(治療的介入による結果)は、死亡率の低下や生活の質(QOL)の向上などであり、これらの評価項目は、真のエンドポイント(true endpoint)と呼ばれます。
しかし、それらを治験の期間内で評価することは難しいため、一般には、短期間で評価できる代用エンドポイント(surrogate endpoint)が採用されます。Surrogateは代理や代用や代替という意味です。
代用エンドポイントは、治療行為に対する評価を短期間で行うための評価項目です。それ自体では臨床上の利益とならなくても、治療上のアウトカムを合理的に予測しうる場合には、プライマリーエンドポイント(主要評価項目)として用いることができます。
がん治療薬の臨床試験の場合、死亡率の低下やQOLの向上が本来求めたいエンドポイント(真のエンドポイント)ですが、短期間で評価するために、奏功率(腫瘍の縮小率)や、増悪や再発が認められるまでの期間(無増悪再発期間、無再発生存期間)などが、代用エンドポイントとして使われています。
代用エンドポイントの結果で有効性が証明されても、真のエンドポイントである死亡率の低下(全生存期間の延長)が証明されている訳ではありません。
前述のように、米国やヨーロッパで承認されたがん治療の新薬の半分くらいが代用エンドポイントの結果で承認されています。これは承認時に全存期間の延長や生活の質の改善が証明されていない抗がん剤が半分を占めることを意味します。
これが市販後に、生存や生活の質の改善が証明されれば問題はありませんが、実際は、代用エンドポイントの結果で承認された薬の多くが、生存や生活の質を改善するエビデンスが得られていないことが明らかになっているのです。
これは代用エンドポイントとして使用されている無増悪生存期間の延長が必ずしも全生存期間の延長を推定できないという原因が大きいようです。無増悪生存期間と全生存期間の相関が低いことは多くの研究で指摘されています。(JAMA Intern Med. 2015;175(8):1389-1398.)
また、無増悪生存期間と全生存期間の相関はがんの種類によっても異なるようです。無増悪生存期間は進行した結腸直腸がんと卵巣がんでは代用エンドポイントとして利用できるが、乳がんと前立腺がんでは代用エンドポイントとして使用できないという結果が報告されています。(Cancer J. 2009 Sep-Oct;15(5):421-5.)
転移のある乳がんでは、奏功率の高い抗がん剤治療のメリットが無いことが多く指摘されています。例えば、米国臨床腫瘍学会(ASCO)のホームページに記載されているChoosing Wisely campaign(「賢い選択を」キャンペーン)の「やってはいけない10のリスト」の7番目には以下のような記述があります。
『転移性乳がんの患者に対して、腫瘍に関連する症状を緩和するために早急に腫瘍を縮小させる必要がある場合以外は、多剤併用の抗がん剤治療を行ってはいけない。転移のある進行乳がん患者に複数の抗がん剤を使用すれば、単剤で治療した場合と比べて腫瘍の増大速度を多少は遅くできるかもしれないが、多剤併用の抗がん剤治療が生存期間を延長するエビデンスは無い。複数の抗がん剤を併用すると副作用が強くなるため、臨床的な利益はむしろ悪化する。』
つまり、進行した乳がんの場合は、奏功率や無増悪生存期間の改善は全生存期間延長と関連しないことが多いようです。
実際に、進行した乳がんの治療薬で無増悪生存期間を主要エンドポイントとして承認された抗がん剤では、全生存期間の延長が証明されていないものが多くあります。
ベバシズマブ(アバスチン)は無増悪生存期間の延長が認められて承認されましたが、全生存期間の延長が証明されなかったため、FDAは承認を取り消しています。
2007年に転移性乳がんに対するベバシズマブとパクリタキセルとの併用で、無増悪生存期間の5.5カ月延長は臨床的に価値があるとみなされて承認されました。しかしその後の臨床試験試験では、無増悪生存期間も全生存期間もどちらも延長を示すデータが示されなかったため、FDAは転移性乳がんへの適応を2011年に取り消しています。(日本では使用されています)
2012年にFDAはエベロリムス(アフィニトール)を転移性乳がんへの使用を承認しましたが、2014年に、臨床試験の追跡調査で生存期間の有意な延長が認められなかったことが明らかになっています。
2015年に新規CDK4/6阻害剤のpalbociclibが、転移性乳がん患者の無増悪生存期間の有意な延長が確認されたので迅速承認されましたが、これも全生存期間を有意に延長する効果は証明されていません。
無増悪生存を臨床試験のエンドポイントとして用いることの妥当性に関して激しい議論が行われています。
無増悪生存期間は、治療中および治療後に疾患の悪化なく生存する期間の長さのことです。一般的には、無増悪生存期間が延びれば、全生存期間も延びるはずですが、必ずしもそうで無いことが明らかになっています。
無増悪生存期間が統計的有意に延長したのに全生存期間は延長しない例は抗がん剤の臨床試験では多数確認されています。その理由は、抗がん剤には強い副作用があるので、がんが増殖を開始するまでの期間が延びても、副作用によって寿命が短縮するので、全生存期間は延びないことが多いのです。
あるいは、抗がん剤使用によって薬剤耐性の悪性度の高いがん細胞が生き残るので、一旦増殖しだすと、抗がん剤を使用しなかった場合より増殖速度が早くなる可能性もあります。
製薬企業のインセンティブは利益であり、利益を増やすには開発中の新薬をできるだけ早く市場に出す必要があります。特許には期限があるからです。時計の針は特許権の期限切れに向かって刻々と進んでいます。特許が切れれば莫大な利益は得られなくなります。
特許権の存続期間は原則として特許出願日から20年で、通常は臨床試験を開始する前に出願するので、臨床試験が終了して薬として認可される頃には特許は10年程度しか残っていません。したがって、臨床試験をできるだけ短縮する必要があります。
生存期間の延長を証明するのには時間がかかるので、奏功率(腫瘍の縮小)や無増悪生存期間が代用の指標として使用され、規制機関(FDA)も新薬を迅速に承認するために、代用エンドポイントで良いとしています。
迅速な承認を求めて圧力をかけているいるのは製薬企業だけでなく、がん患者からの要求もあります。
そのような事情が重なって、生存期間を延ばせないのに、奏功率(腫瘍縮小率)や無増悪生存期間の延長を示して、多くの抗がん剤が承認されているという状況になっているようです。
その結果、延命効果が証明されていない高額な抗がん剤が使用されるようになっています。
| « 702)ビタミンD... | 704)精神的ス... » |