がんの予防や治療における漢方治療の存在意義を考察しています。がん治療に役立つ情報も紹介しています。
「漢方がん治療」を考える
カレンダー
| 2025年2月 | ||||||||
| 日 | 月 | 火 | 水 | 木 | 金 | 土 | ||
| 1 | ||||||||
| 2 | 3 | 4 | 5 | 6 | 7 | 8 | ||
| 9 | 10 | 11 | 12 | 13 | 14 | 15 | ||
| 16 | 17 | 18 | 19 | 20 | 21 | 22 | ||
| 23 | 24 | 25 | 26 | 27 | 28 | |||
|
||||||||
goo ブログ
過去の記事
カテゴリ
最新の投稿
最新のコメント
最新のトラックバック
ブックマーク
プロフィール
検索
gooおすすめリンク



| URLをメールで送信する | |
| (for PC & MOBILE) | |
398)糖質制限は抗がん剤治療による老化促進を予防する
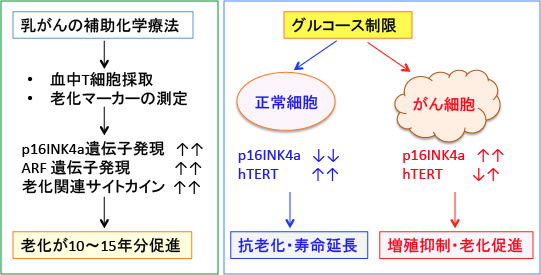
図:(左)乳がんの補助化学療法を受けた患者の末梢血T細胞の老化関連マーカーを測定すると、細胞の老化に伴って細胞内発現量が多くなるp16INK4aとARF遺伝子の発現と老化関連のサイトカイン(VEGFAとMCP1)の増加が認められた。その増加の程度は細胞の老化が10~15年間分の促進に相当していた。
(右)正常なヒト胎児肺線維芽細胞(正常細胞)と、この細胞にがん遺伝子を導入して作成した不死化細胞(がん細胞)は、グルコース制限に対して相反する反応を示した。正常細胞ではグルコース制限によってp16INK4a発現は減少しhTERT発現は増加した。がん細胞では逆の応答を示し、グルコース制限によってp16INK4a発現は増加しhTERT発現は減少した。
p16INK4aは細胞周期を止め細胞老化を促進する作用があり、hTERT(human telomerase reverse transcriptase; ヒト・テロメラーゼ逆転写酵素)はテロメアの短縮を防いで細胞分裂能の喪失を防ぐ作用がある。したがって、グルコース制限は正常細胞に対しては老化抑制・寿命延長の効果を示し、がん細胞に対しては増殖抑制・老化促進作用を示すことになる。
398)糖質制限は抗がん剤治療による老化促進を予防する
【老化のマーカーとしてのp16INK4a】
生物は成長が終了した後(人間では20~30歳以降)、加齢(aging)とともに細胞や組織の機能が低下していきます。このような加齢に伴う生理機能の低下を老化と言います。
体の組織は組織幹細胞が細胞分裂によって細胞を補うことによって正常に維持されますが、加齢とともに組織幹細胞も老化によって次第に再性能(細胞分裂能)を失い、やがて死滅し、数が減っていきます。その結果、組織の細胞量が減少し機能が低下していきます。
この老化の速度は個人差があり、遺伝的要因や生活要因や環境要因によっても影響をうけます。
老化の程度や速度を評価するには「老化のマーカー」が必要です。これは、老化に伴って細胞内量が増えるか減るような(細胞の老化の程度と相関するような)細胞内物質が候補になります。
例えば、形態学的な細胞老化のマーカーとしてリポフスチンがあります。リポフスチン(lipofuscin)は、細胞内の不飽和脂肪酸の過酸化によってリソソーム内に形成される不溶性色素で、加齢性色素と呼ばれます。老化した肝細胞や心筋細胞でみられます。
生化学的なマーカーとしては細胞老化関連ベータ・ガラクトシダーゼ(senescence-associated β-galactosidase)があります。老化した細胞はこの酵素の活性が高くなっており、X-galを使って老化細胞を染色することができます。
老化の分子マーカーとしてよく使われているのがp16INK4aというタンパク質です。P16INK4aは細胞の老化にともなって指数関数的に増加することが知られています(細胞内のp16INK4aの量は16.7年で2倍になるという報告があり、この倍加時間で倍々になっていきます)。つまり、細胞内のp16INK4aの発現量が多いほど、その細胞は老化していると評価されています。
p16INK4aタンパク質はサイクリン依存性期キナーゼ(cyclin dependent kinase: CDK)4と6(CDK4とCDK6)に結合する分子量が約16000ダルトン(16kDa)のタンパク質です。
p16はCDK4とCDK6に結合することによって、CDK4とCDK6がサイクリンD(cyclin D1, D2, D3)と結合することを阻害します。その結果、CDK4とCDK6は活性化できません。CDK4とCDK6は細胞が分裂をスタートするときに必須の働きを行うので、このサイクリン依存性期キナーゼが阻害されると細胞は分裂できません。
細胞が分裂して数を増やしていくとき、細胞周期は4 つの段階に分けられます。すなわち、DNA複製前のG1(Gap1) 期、DNA複製期(S期)、細胞分裂前のG2(Gap2)期、および最後の細胞分裂期(M) 期に分けられます。増殖を休止した状態の細胞はG0期にあると定義されます。
細胞周期がG1期からS期に移行するときがん抑制遺伝子のRBタンパク質がサイクリン依存性キナーゼ(CDK)でリン酸化されることが重要です。RBタンパク質がサイクリン依存性キナーゼでリン酸化されると転写因子のE2Fと結合できなくなり、フリーになったE2Fは増殖に関連する遺伝子の発現を促進して細胞周期を回します。
(RBタンパク質はE2Fに結合してE2Fの転写活性を阻害している)
サイクリン依存性キナーゼ(CDK)はサイクリン依存性キナーゼ阻害因子というタンパク質によって機能が阻害されます。このサイクリン依存性キナーゼ阻害因子にはp16INK4a やp21Waf1/Cip1/Sdi1などのタンパク質が知られています。
(CDKにCDK阻害因子が結合することによってCDKとサイクリンの結合を阻害する。CDKはサイクリン依存性のキナーゼなので、サイクリンに結合できないと活性化できない)(下図)
細胞の老化というのは、細胞が不可逆的に分裂を停止する(細胞分裂能を喪失する)事です。細胞にダメージが蓄積して老化してくると、サイクリン依存性キナーゼ阻害因子のp16INK4aの発現量が増えてきて、細胞周期が回らないようにすると考えられます。したがって、老化した細胞ほどp16INK4aの発現量が増えており、細胞老化のマーカーとなるのです。

図:G1期にRBタンパクはE2Fという転写因子に結合して、E2Fの活性を抑えている。E2Fは転写因子で細胞の増殖にとって重要な多くの遺伝子類の発現を亢進する。したがって、RBが結合してE2Fの活性を抑えていると、細胞は増殖サイクルに入れない。しかし、サイクリン依存性キナーゼ(Cdk4あるいはCdk6)とサイクリンDの複合体によってRBがリン酸化されるとRBタンパクはE2Fから解離し、E2Fが活性な転写因子となって増殖関連遺伝子の発現を引き起こすので、細胞はDNA複製を開始して増殖サイクルを回し出す。
p16INK4aはサイクリン依存性キナーゼのCdk4とCdk6と結合することによって阻害する。Cdk4/6の活性が阻害されると、RBはリン酸化されないので、細胞周期はストップした状態に維持される。老化した細胞(分裂能を喪失した細胞)ではp16INK4aの発現量が増えており、p16INK4aは細胞の老化のマーカーとして知られている。
【乳がんの補助化学療法は老化を10年~15年間進める?】
がんがある程度進行している場合は、外科切除の後(場合によっては切除前)に抗がん剤治療が行われます。これを補助化学療法(Adjuvant chemotherapy)といいます。
補助化学療法は、正常細胞へのダメージによって、内分泌系の異常、認知力低下、心血管系疾患、神経や筋肉系の異常、別のがんの発生(2次がん)など、長期的なデメリットを有する可能性が指摘されていますが、短期的には生存率を高める効果があるため、メリットの方が高いという判断で、標準的に行われています。
補助化学療法の長期的な副作用や後遺症やデメリットについてはあまり重視されていませんが、実際は極めて重要であり、それを防ぐ方法を知ることはがんサバイバーには必要です。
例えば、ホジキンリンパ腫は30年前は治癒しない腫瘍でしたが、現在では強力な抗がん剤治療と放射線治療の組合せによって7~8割が治るようになっています。
しかし、このような治療を受けた患者の25年間の2次がん(乳がん、肺がん、消化器がん、甲状腺がん)の発症率は22%におよぶという報告があります。
20歳以下でホジキン病の治療(抗がん剤+放射線治療)を受けた女性患者が10年間で乳がんを発症する率は40%という報告もあります。
白血病や心筋傷害は数%の率で発症しています。
その他、多くのがんで強力な抗がん剤治療や放射線治療で治療成績は向上していますが、強力な治療ほど長期的な副作用(認知力の低下、心機能低下、2次がん、など)が問題になっています。このような副作用や後遺症を軽減し予防することが重要です。
体や細胞の老化を促進したり、寿命を短縮する可能性も指摘されています。
乳がんの補助化学療法を受けた患者さんは老化が10年~15年程度進む可能性を示唆する報告があります。以下のような論文があります。
Effect of Cytotoxic Chemotherapy on Markers of Molecular Age in Patients With Breast Cancer (乳がん患者における寿命のマーカーに対する細胞傷害性抗がん剤の作用)
J Natl Cancer Inst. 2014 Apr;106(4):dju057. doi: 10.1093/jnci/dju057. Epub 2014 Mar 28.
【要旨】
研究の背景:p16INK4aを発現している老化した細胞は生体の加齢とともに増え、老化関連疾患の発症に関与している。細胞傷害性の抗がん剤が生物学的な老化を促進するかどうかを明らかにする目的で、補助化学療法を受けた乳がん患者において、p16INK4aおよびその他の老化関連マーカーを測定した。
方法:ステージIからIIIの乳がん患者33名を対象に、①アントラサイクリンをベースにした抗がん剤治療の前、②抗がん剤治療の終了直後、③抗がん剤治療終了の3ヶ月後、④抗がん剤治療終了の12ヶ月後の4つの時点において、血液と臨床的情報を収集した。
CD3陽性T細胞を採取して、細胞老化のマーカーであるp16INK4αとARF遺伝子のmRNAの発現量、テロメアの長さ、老化関連のサイトカインの定量を行った。
176例の乳がん(治療後平均期間が3.4年、39%が抗がん剤投与を受けている)の別の集団を対象にした検討も実施した。
結果:前向きに解析した患者において、p16INK4αとARFのmRNAの発現量は抗がん剤治療直後から上昇し、治療後12ヶ月経過した時点でも高い値を維持していた。
P16INK4αの上昇率の中央値は75%であり、この値は、生物学的な加齢に換算すると14.7年分の加齢に相当する。ARFの発現量も同程度に増加した。
P16INK4αとARFの発現量の増加は、抗がん剤投与量と血液学的毒性の程度と相関していた。
2種類の老化関連のサイトカイン(VEGFAとMCP1)は補助化学療法によって持続的に上昇した。
テロメアの長さは抗がん剤治療で変化は認めなかった。
別の集団での検討では、抗がん剤治療を受けた患者ではp16INK4αの発現量が増加し、それは10.4年の加齢促進に相当する増加であった。
結論:乳がんに対する補助化学療法は、生体内における細胞の老化を促進し、造血組織の加齢を促進する作用がある。
この論文では、乳がん患者の2つの集団(コホート)で検討しています。一つは33人の乳がん患者を経時的に追跡する前向き研究で、もう一つは治療後平均3.4年が経過している乳がんサバイバーのコホートで、このコホートにおいて補助化学療法を受けた人と受けていない人で比較しています。
末梢血のT細胞のP16INK4aやARF遺伝子の発現量は加齢とともに増加することが知られており、この遺伝子発現量(mRNA量)を老化のマーカーとして検討すると、乳がんで術前あるいは術後の補助化学療法は、加齢を10~15年分促進する(つまり、寿命を10~15年間ほど短縮する)というデータが得られたということです。
ARFとはAlternative Reading Frameの意味で、p16INK4aとARF遺伝子は同じ遺伝子領域から転写されますが、p16INK4とは違ったコーディングフレーム(cording frame)を利用して作られるのがARFで、これも老化のマーカーです。
老化関連の遺伝子のp16INK4aとARFと2種類の老化関連のサイトカイン(VEGFAとMCP1)が抗がん剤治療によって増えているので、抗がん剤治療は老化を促進する、しかも、増えた量から換算すると、10年から15年に相当する年月の老化が促進していたという結論です。
しかも、老化のマーカーは、前向き研究のコホートでは抗がん剤治療直後から上昇し、抗がん剤治療終了12ヶ月後も上昇しており、治療後平均3.4年が経過したコホートでも上昇が認められるので、抗がん剤治療による老化促進の影響は数年あるいは永久に持続する可能性が示唆されると言っています。
ただし、末梢血のTリンパ球だけの検討なので、体全体の寿命をそれだけ短縮するかどうかは不明です。造血組織は抗がん剤感受性が高くダメージを受けやすいので、このような顕著は差が出た可能性はあります。体全体では数年間かあるいはもっと短いかもしれません。
しかし、細胞傷害性の抗がん剤治療が細胞や体の老化を促進することは十分に納得できると思います。
【糖質制限は正常細胞のp16INK4αの発現を抑制して老化速度を遅くする】
p16INK4αが増えた細胞は老化した細胞で、この量があるレベルを超えると細胞分裂能を喪失すると考えられます。したがって、細胞内のp16INK4αの発現量は「細胞の老化の程度」と評価するマーカーになります。一方、p16INK4αの遺伝子発現の抑制は細胞の寿命を伸ばすことにつながります。
以下のような論文があります。
p16(INK4a) suppression by glucose restriction contributes to human cellular lifespan extension through SIRT1-mediated epigenetic and genetic mechanisms.(グルコース制限によるp16INK4a遺伝子発現の抑制は、SIRT1-介在性のエピジェネティックあるいはジェネティックなメカニズムでヒト細胞の寿命延長に貢献する)PLoS One. 2011 Feb 24;6(2):e17421. doi: 10.1371/journal.pone.0017421.
【要旨】
カロリー制限が様々な動物において寿命を延長することが確認されているが、そのメカニズムについては十分に解明されていない。
培養液中のグルコースの濃度を減らすことによってカロリー制限と類似の状況を作り出す培養細胞の実験モデルを使って、細胞分子レベルでカロリー制限(グルコース制限)の作用メカニズムを検討した。
正常のヒト肺線維芽細胞(WI-38, IMR-90, MRC-5)においては、正常のグルコース濃度で培養した細胞と比べて、グルコースの量を減らした(グルコース制限)培養液で培養した細胞では、老化速度が抑制され、細胞の寿命が顕著に延長した。
さらに、検討した全ての細胞株において、グルコース制限は老化関連遺伝子として知られているp16INK4aの発現量を減少させた。
グルコース制限を行っている細胞(老化が抑制されている細胞)にp16遺伝子を過剰発現させると、細胞分裂が早期に停止し細胞老化が促進された。この結果は、グルコース制限による細胞寿命の延長にp16が重要な役割を担っていることを示している。
P16の発現抑制は、グルコース制限によってヒストン・アセチル化によるクロマチン再構成とp16遺伝子のプロモーター領域のメチル化が関与していた。
グルコース制限はNAD依存性のヒストン・脱アセチル化酵素であるSIRT1(サーチュイン1)の発現を増やした。SIRT1はカロリー制限による寿命延長と関連していることが知られている。
SIRT1の発現亢進はAkt/p70S6K1シグナル伝達系の活性を促進する。SIRT1遺伝子を欠損させた細胞では、グルコース制限によるp16発現の抑制とAkt/p70S6K1シグナル伝達系の活性化は起こらなかった。
これらの結果は、SIRT1はヒストンの脱アセチル化による直接作用とAkt/p70S6K1シグナル伝達系を介する間接的な作用によってp16遺伝子の発現を抑制することを示唆している。
複雑なメカニズムですが、簡単にまとめると「正常細胞に対してグルコース制限はp16INK4aの発現を抑制して、細胞の老化を遅らせる」ということです。そのメカニズムとしてSIRT1やヒストンアセチル化などが関与しているという話です。
【糖質制限はがん細胞のp16INK4αの発現を亢進して増殖を抑制する】
前述のように、補助化学療法は再発予防の目的のためにはメリットはありますが、正常細胞のダメージから、様々な副作用を引き起こし、長期的にも老化を促進し、寿命を短縮する可能性があります。
老化促進・寿命短縮という抗がん剤治療の長期的なデメリットを防ぐ方法としてカロリー制限や糖質制限が有効の可能性があります。
前述のように老化のマーカーであるp16INK4aを指標にした研究で糖質制限が正常細胞のp16INK4aの発現量を減少させて老化速度を遅くするという報告があります。
さらに、グルコース制限はがん細胞に対しては、正常細胞とは逆の応答をするという報告があります。以下のような論文があります。
Glucose restriction can extend normal cell lifespan and impair precancerous cell growth through epigenetic control of hTERT and p16 expression.(糖質制限はhTERT遺伝子とp16遺伝子の発現をエピジェネシスによる制御を介して正常細胞の寿命を延ばし、前がん細胞の増殖を抑制する)FASEB J. 24(5): 1442-1453, 2010年
【要旨】
がん細胞はグルコースの取込みと代謝が亢進しており、そのためグルコースの制限に対して正常細胞に比べて増殖や生存に対する影響が大きい。しかしながら、正常細胞とがん細胞のグルコース制限に対する影響の違いに関する分子メカニズムは十分に解明されていない。
正常な胎児肺線維芽細胞WI-38と、WI-38にがん遺伝子を導入して不死化したWI-38/S細胞を用いてグルコース制限に対する影響を検討した。その結果、不死化したWI-38/Sではグルコース制限によって増殖阻害とアポトーシスの誘導が認められたが、正常細胞のWI-38ではグルコース制限は寿命を延ばした。
さらに、WI-38/S細胞では、グルコース制限によってhTERT(human telomerase reverse transcriptase; ヒト・テロメラーゼ逆転写酵素)の発現が減少し、がん抑制遺伝子のp16INK4aの発現が増加した。
正常細胞のWI-38細胞では、hTERT遺伝子とp16遺伝子の発現に対するグルコース制限による影響は逆であった。(WI-38細胞では、グルコース制限でhTERT遺伝子の発現は増加し、p16INK4a遺伝子の発現や減少した)
このようなWI-38とWI-38/S細胞の遺伝子発現の違いは、hTERT遺伝子とp16遺伝子のプロモーター領域におけるDNAのメチル化やクロマチン再構成(chromatin remodeling)におけるグルコース制限による変化がWI-38とWI-38/Sとで異なることが関与していると思われた。
さらに、グルコース制限によるhTERT遺伝子とp16遺伝子の発現量の変化はWI-38の方がWI-38/S細胞より明らかであり、これはエネルギー低下によるストレスで誘導される遺伝子のエピジェネシスによる制御が正常細胞と前がん細胞で異なっていることを示唆している。
以上の結果から、食事による遺伝子のエピジェネシスによる制御が、がんの治療法と抗老化の手段となりうることを示している。
真核生物の染色体には端っこにテロメアと呼ばれる配列があり、細胞が分裂するたびにこのテロメアが減っていきます。このテロメアがある程度短くなると細胞は分裂できなくなります。つまり細胞分裂能の喪失(=老化)ということになります。
短くなったテロメアをもう一度伸ばしてくれる酵素が「テロメラーゼ」でこのタンパク質をコードしている遺伝子がhTERT遺伝子です。hTERT遺伝子の発現が増加するというのは、細胞の分裂する回数を増やすことができます。
普通の正常細胞ではテロメラーゼはほとんど発現していないため、細胞分裂の回数に限界があります。
一方、多くのがん細胞では、テロメラーゼが発現し、活性が亢進しています。その結果、無限に増殖できることになります。
この論文の結果では、がん細胞ではグルコース制限によってhTERTの発現が減少し、がん抑制遺伝子のp16INK4aの発現が増加し(=細胞が老化の方向に向かう変化)、正常細胞では逆に、グルコース制限でhTERT遺伝子の発現は増加し、p16INK4a遺伝子の発現が減少しました(=細胞が老化を抑える変化)。
培養細胞での検討ですが、糖質制限はがん細胞の老化を促進し、正常細胞の老化を抑制する可能性を示しています。
正常細胞の培養液にグルコースの量を増やすと老化が促進されることが報告されています。逆にがん細胞の場合は、グルコールの取込みと利用が亢進しているので、グルコースの量を増やすとがん細胞の増殖が促進されます。
つまり、正常細胞とがん細胞では、グルコース濃度に対する対応が全く異なります。
生体でも、高血糖はがん細胞の増殖を促進し、正常細胞の老化を促進します。したがって、糖質制限はがん細胞の増殖を抑制し、正常細胞の老化を抑制することになります。
がんの治療や再発予防やがんサバイバーにおける長期的な副作用(老化促進や2次がんなど)の予防に糖質制限が役立つことを示唆する研究結果だと思います。


画像をクリックするとサイトに移行します。
| « 397) 漢方がん... | 399)水素ガス... » |