がんの予防や治療における漢方治療の存在意義を考察しています。がん治療に役立つ情報も紹介しています。
「漢方がん治療」を考える
カレンダー
| 2025年4月 | ||||||||
| 日 | 月 | 火 | 水 | 木 | 金 | 土 | ||
| 1 | 2 | 3 | 4 | 5 | ||||
| 6 | 7 | 8 | 9 | 10 | 11 | 12 | ||
| 13 | 14 | 15 | 16 | 17 | 18 | 19 | ||
| 20 | 21 | 22 | 23 | 24 | 25 | 26 | ||
| 27 | 28 | 29 | 30 | |||||
|
||||||||
goo ブログ
過去の記事
カテゴリ
最新の投稿
最新のコメント
最新のトラックバック
ブックマーク
プロフィール
検索
gooおすすめリンク




| URLをメールで送信する | |
| (for PC & MOBILE) | |
633)NADを枯渇させるがん治療:ジクロフェナク+高濃度ビタミンC点滴+半枝蓮+メトホルミン

図:がん細胞ではグルコース・トランスポーター1(GLUT1)からグルコースの取り込みが増えている(①)。解糖系では1分子のグルコースから2分子のピルビン酸、2分子のATP、2分子のNADH + H+が作られる(②)。乳酸脱水素酵素は、NADH + H+を還元剤として用いてピルビン酸を還元して乳酸にする(③)。この乳酸発酵によってNAD+を再生することによって解糖系での代謝が続けられる(④)。ジクロフェナクは乳酸脱水素酵素を阻害する(⑤)。抗がん剤や放射線や高濃度ビタミンCや半枝蓮はDNAにダメージを与え、ポリ(ADP-リボース)ポリメラーゼ(PARP)を活性化する(⑥)。PARPの活性化はニコチンアミドアデニンジヌクレオチド(NAD+)を減少する(⑦)。メトホルミンはミトコンドリアの呼吸酵素複合体IとmGDP(ミトコンドリア・グリセロールリン酸脱水素酵素)を阻害し(⑧)、NADHの酸化を阻害するのでNAD+が減少する(⑨)。これらによって、細胞内のNAD+が枯渇すると、がん細胞内のATPが枯渇してがん細胞は死滅する(⑩)。
633)NADを枯渇させるがん治療:ジクロフェナク+高濃度ビタミンC点滴+半枝蓮+メトホルミン
【がん細胞はなぜ乳酸産生が増えているのか】
がん細胞の代謝の特徴は、酸素が十分にあってもミトコンドリアでの酸化的リン酸化によるエネルギー(ATP)産生が抑制され、酸素を使わないグルコースの分解(解糖系)が亢進していることです。そのため、グルコース(ブドウ糖)の取込みが増え、乳酸の産生が増えています。これをワールブルグ効果あるいは好気性解糖と言います。
解糖系でできたピルビン酸は、嫌気的条件(酸素が無い状態)では乳酸に変換されます。
グルコースからピルビン酸まで分解したあと(この過程を解糖という)、酸素があればTCA回路(クエン酸回路)と電子伝達系による酸化的リン酸化によってATPを生成しますが、酸素が無い場合はピルビン酸からさらに乳酸に変換されます。ピルビン酸を乳酸に変換する酵素が乳酸脱水素酵素(Lactate Dehydrogenase; LDH)です。(下図)。

図:グルコースが解糖系でピルビン酸まで分解されたあと、酸素があればミトコンドリアでピルビン酸脱水素酵素(Pyruvate Dehydrogenase; PDH)によってアセチルCoAに変換されてTCA回路でさらに代謝され、電子伝達系(呼吸鎖)でATPが産生される。酸素が無い条件では、ピルビン酸は乳酸脱水素酵素(Lactate Dehydrogenase; LDH)によって乳酸に変換される。がん細胞では、LDHの活性が亢進し、PDHの活性は抑制されており、酸素が十分に使える状況でも、ミトコンドリアでの酸素呼吸(酸化的リン酸化)は抑制され、乳酸産生が亢進している。
がん細胞の場合は、酸素が十分にあっても、ミトコンドリアでの酸化的リン酸化を抑制しているので、乳酸の方に行きます。
なぜ、ピルビン酸で止まらないで乳酸に変換されるかというと、その理由は、解糖系で還元されたNADH(還元型ニコチンアミドジヌクレオチド)を酸化型のNAD+に戻すためです。NAD+が枯渇すると解糖系が進行しなくなります。(下図)

図:解糖系では1分子のグルコースから2分子のピルビン酸、2分子のATP、2分子のNADH + H+が作られる。乳酸発酵では、NADH + H+を還元剤として用いてピルビン酸を還元して乳酸にする。この乳酸発酵によってNAD+を再生することによって酸素を使わないATP産生(解糖)が続けられる。その結果、乳酸が多く産生される。
解糖系でのグルコースからピルビン酸への代謝で、1分子のグルコースから2分子のATPを産生できます。乳酸発酵によって酸化型NAD(NAD+)を再生することによって、がん細胞は無酸素条件下で生きてはいけるのです。
人間を含めて全ての哺乳動物は無酸素の環境では長くは生存できません。しかし、少し下等な生物では無酸素でも長く生きられるものが存在します。
淡水にすむカメ(亀)は無酸素で長く生きることができます。
淡水産カメ、特にニシキガメ(Chrysemys picta)は、空気を呼吸する脊椎動物の中で、最も長く無酸素に耐えることができます。このカメは3℃の無酸素状態の水中で5ヶ月間生存できることが実験で示されています。
ニシキガメ(Chrysemys picta)は米国でよく見られる淡水産のカメで、頭頂部に鮮やかな黄色の斑紋や背甲の縁に赤い模様があります。

カメの骨格の特徴は甲羅です。甲羅の内部は脊椎骨、肩胛骨、肋骨、胸骨などが互いに密着して箱のような構造をしています。嫌気性解糖で産生された乳酸を甲羅の内部で貯蔵して、炭酸塩で緩衝させるメカニズムが進化して、乳酸蓄積の有害性を軽減できるので、カメは無酸素の状態で長く生きられると考えられています。(Comp Biochem Physiol A Mol Integr Physiol. 2000 Mar;125(3):299-315.)
カメの寿命が100年以上と長いのは酸素を使わないからだという意見もあります。
一般に動物は、単位体重当たりの酸素消費と寿命は反比例すると言われています。つまり、酸素消費が少ないほど長生きできます。
がん細胞も、乳酸産生を増やすことによって、酸素を使わない方法でATP産生を行い、長く生存できるのです。
【乳酸脱水素酵素の阻害やDNAの損傷はNAD+を枯渇する】
ニコチン酸とニコチン酸アミドは総称してナイアシン(Niacin) 、あるいはビタミンB3とも言います。水溶性ビタミンのビタミンB複合体の一つで、糖質や脂質やタンパク質の代謝に不可欠です。

ナイアシンは電子伝達体のニコチンアミドアデニンジヌクレオチド(NAD) やニコチンアミドアデニンジヌクレオチドリン酸(NADP) に変換され、酸化還元反応(電子が供与体分子から受容体分子に転移する反応) に関与する酵素の補酵素として機能しています。また、NADが基質として利用されるADP-リボシル化反応にも関与しています。

前述のように、解糖系で還元されたNADH(還元型ニコチンアミドジヌクレオチド)を酸化型のNAD+に戻すために、乳酸脱水素酵素でピルビン酸を乳酸に変換しています。従って、乳酸脱水素酵素を阻害して乳酸発酵を阻害するとNAD+が枯渇して解糖系が進行しなくなります。
また、DNA切断もNAD+を消費して枯渇させます。
ADP-リボシル化は細胞内で起こるたんぱく質の修飾で、NADのADPリボース部分がポリADPリボースポリメラーゼによりたんぱく質分子に付加されます。この反応はDNA修復やテロメアの維持において重要な役割を果たしています。
ポリ(ADP-リボース)ポリメラーゼ(PARP)は細胞内に多く存在するタンパク質で、核DNAに生じた一本鎖切断端を認識してDNAに結合します。核DNAに結合したPARPは活性化され、ニコチンアミドアデニンジヌクレオチド(NAD+)を基質としてPARP自身やヒストンやDNA修復関連タンパク質にADP-リボースを付加し、ポリ-ADP-リボシル化を引き起こします。このポリ-ADPリボシル化はDNA修復するシグナルとなります。
通常、ポリ-ADP-リボシル化はDNA修復反応を活性化しますが、過度のPARPの活性化はNAD+とATPの枯渇、さらにミトコンドリアに局在するアポトーシス誘導因子(AIF)の切断を誘導します。
切断されて細胞質に放出されたAIFはミトコンドリアに局在していたエンドヌクレアーゼGとともに核に移行し,核DNAの断片化を引き起こし,細胞死を誘導します。

図:ポリ(ADP-リボース)ポリメラーゼ(PARP)は、核DNAに生じた一本鎖切断端(①)を認識してZn-finger domain(Zf)部分でDNAに結合する(②)。核DNAに結合したPARPは活性化され、ニコチンアミドアデニンジヌクレオチド(NAD+)を基質としてPARP自身やDNA修復関連タンパク質にADP-リボースを付加し(③)、ポリ-ADP-リボシル化を引き起こし(④)、他の修復タンパク質をリクルートし(⑤)、DNAを修復する(⑥)。過度のDNA損傷やPARPの過剰な活性化(⑦)は、NAD+とATPを枯渇し(⑧)、細胞死を誘導する(⑨)。
【がん細胞では乳酸脱水素酵素Aの発現と活性が亢進している】
細胞内に取り込まれたグルコースは、解糖系で代謝されてピルビン酸まで分解されます。その後、正常細胞では、酸素が使える状況ではピルビン酸はミトコンドリアに入り、ピルビン酸脱水素酵素によってアセチルCoAに変換されてTCA回路でさらに代謝されます。低酸素の状況では、ピルビン酸は細胞質で乳酸脱水素酵素(Lactate Dehydrogenase: LDH)によって乳酸に変換されます。
一方がん細胞では、酸素が十分に使える状況でも、ミトコンドリアでの酸素呼吸(酸化的リン酸化)が抑制されています。その理由としての一つは、ピルビン酸脱水素酵素キナーゼによってピルビン酸脱水素酵素(PDH)の活性が抑制されているからです。その結果、ピルビン酸はミトコンドリアで代謝されずに、細胞質内の乳酸脱水素酵素によって乳酸に変換されます。

図:グルコースが解糖系でピルビン酸まで分解されたあと、酸素があればミトコンドリアでピルビン酸脱水素酵素(Pyruvate Dehydrogenase; PDH)によってアセチルCoAに変換されてTCA回路でさらに代謝され、電子伝達系(呼吸鎖)でATPが産生される。がん細胞ではピルビン酸脱水素酵素キナーゼ(PDHK)の活性が亢進してPDHを阻害している(①)。その結果、ミトコンドリアでの酸化的リン酸化によるATP産生が抑制されている(②)。一方、がん細胞ではグルコースの取り込みと解答系が亢進し(③)、さらに乳酸脱水素酵素(Lactate Dehydrogenase; LDH)の活性が亢進して、ピルビン酸の多くは乳酸に変換される(④)。がん細胞では、LDHの活性が亢進し、PDHの活性は抑制されており、酸素が十分に使える状況でも、ミトコンドリアでの酸素呼吸(酸化的リン酸化)は抑制され、乳酸産生が亢進している。
LDHにはLDH-AとLDH-Bの2つのサブタイプがあります。
LDH-Aは骨格筋タイプあるいはLDH-Mとも言い、ピルビン酸から乳酸の変換に適しています。骨格筋では通常は有酸素でミトコンドリアでの代謝が中心ですが、短距離ダッシュのような無酸素での運動では骨格筋で嫌気性解糖によるエネルギー産生が起こるのでLDH-Aが必要になります。
一方、LDH-Bは心臓タイプあるいはLDH-Hとも言い、乳酸からピルビン酸の変換に適しています。心臓では、血中の乳酸もエネルギー源に利用するので、乳酸からピルビン酸に変換するLDH-Bが必要になります。

図:乳酸脱水素酵素(LDH)はピルビン酸から乳酸への変換を促進するLDH-Aと、乳酸からピルビン酸への変換を促進するLDH-Bの2つのタイプがある。
LDH-Aはがん治療のターゲットになります。それは好気性解糖(ワールブルグ効果)が亢進しているがん細胞ではLDH-Aの発現が亢進しているのに対して、正常細胞では骨格筋にしか発現していないためです。LDH-Aは骨格筋で嫌気性解糖を行うときしか必要ないので、短距離ダッシュのように無酸素の運動をしなければ、LDH-Aは正常細胞では無くても構わないと言えます。実際にLDH-Aの遺伝子が欠損していても、大した異常は起こらないことが報告されています。
多くのがん細胞でLDH-Aの発現亢進が認められています。LDH-Aは低酸素誘導因子-1(HIF-1)によって発現が誘導されます。がん細胞ではHIF-1の発現が亢進しており、LDH-Aの発現が亢進しています。
LDHの血中濃度が高いほど予後不良というデータが多数報告されています。
がん細胞のLDHの活性が高いほど、乳酸とプロトンの産生が増加しており、がん組織の酸性化が亢進し、このような状況はがん細胞の浸潤性を亢進します。
つまり、がん細胞のLDH活性亢進→がん組織の酸性化亢進→がん細胞の浸潤性亢進→その結果、予後不良になる、というストーリーです。
進行がんの患者における血中のLDHの多くは死滅したがん細胞に由来するので、血中のLDHの値が高いほど、がんの進行が進んでいるということになります。以下のような報告があります。
Prognostic role of lactate dehydrogenase in solid tumors: a systematic review and meta-analysis of 76 studies.(固形がんにおける乳酸脱水素酵素と予後との関係:76件のメタ解析と系統的レヴュー)Acta Oncol.2015 Jul;54(7):961-70.
【要旨】
背景:がん細胞では、代謝が好気性解糖にシフトし、エネルギー源としてグルコースの取込みが亢進し、乳酸の産生が増えている。乳酸脱水素酵素(LDH)がピルビン酸から乳酸への変換を触媒する。進行したがんや血液腫瘍では血清中のLDHの値は上昇している。固形がんにおける血清中のLDHの値と予後との関連を検討した。
対象と方法:固形がんにおける血清LDH値と予後との関連を検討した論文を検索し、メタ解析を行った。
結果:76件の研究の22,882人が対象で、主に進行したがん患者であった。LDH値が高いほど全死因死亡率が高かった(ハザード比1.7: 95%信頼区間1.62-1.79)
LDHと予後との関連は、腎臓がん、メラノーマ、胃がん、前立腺がん、鼻咽頭がん、肺がんで強く認めた。
結論:固形がん、特にメラノーマ、前立腺がん、腎臓がんにおいて、血清LDH値の高値は予後不良との関連を認めた。転移性のがんにおいて、血清LDH値は予後を推定する安価で有用なマーカーとして使用できる。
乳酸脱水素酵素(LDH)はほとんどの細胞に含まれていますが、特にLDHが多く含まれている臓器は、肝臓、腎臓、心筋、骨格筋、赤血球などです。
肝臓や心臓などの臓器に異常が起き細胞が死滅すると血液中にLDHが流れ出して、LDH値が高い状態を示すようになります。
がん以外では、急性肝炎や心筋梗塞、うっ血性心不全、進行性筋ジストロフィー症、溶血性貧血などのときに著しくLDHが増加します。
多くのがんでLDHは上昇します。がん細胞は正常細胞よりLDHの発現量が多いからです。がんが大きくなると、がん組織の中で死滅する細胞も増えるので、血中にLDHが流出して血清LDH値が高くなります。
したがって、血清LDHの値はがんの進行のマーカーとなり、予後とも関連することになります。
【ジクロフェナクのLDH-A阻害作用】
ジクロフェナクは商品名がボルタレンというポピュラーな消炎鎮痛剤です。以下のような報告があります。
Diclofenac inhibits lactate formation and efficiently counteracts local immune suppression in a murine glioma model.(ジクロフェナクはマウスのグリオーマの実験モデルにおいて、乳酸産生を阻害し、免疫抑制を効率的に解除する)Int J Cancer 132(4): 843-853, 2013年
【要旨】
悪性のグリオーマ(神経膠腫)のように増殖活性が高い腫瘍組織で増加している乳酸産生は生存率の低下(予後不良)と密接に関連し、さらに腫瘍局所の免疫応答の抑制を引き起こす要因となっている。
この論文では、培養細胞を用いたvi vitroの実験系において、ジクロフェナクが毒性を引き起こさないレベルの濃度でマウスのグリオーマ細胞における乳酸産生量を顕著に減少させることを示し、そのメカニズムとして乳酸脱水素酵素-A(lactate dehydrogenase-A)の発現を阻害する作用との関連を報告した。乳酸産生の減少と同時に、用量依存的な細胞増殖の阻害とG2/Mチャックポイントでの細胞周期の停止も認められた。
マウスの骨髄由来樹状細胞とグリオーマ細胞を一緒に培養する実験系において、骨髄由来樹状細胞をToll様受容体をR848で刺激する際にジクロフェナクを添加すると、樹状細胞からのIL-12の産生分泌が亢進し、IL-10(免疫抑制性に作用するサイトカイン)の産生は抑制された。この際、グリオーマ細胞の乳酸産生は減少し、STAT-3(シグナル伝達兼転写活性化因子3:Signal Transducers and Activator of Transcription-3)のリン酸化が阻害された。
グリオーマの移植腫瘍を用いた動物実験では、ジクロフェナクを投与すると腫瘍内の乳酸濃度は低下し、グリオーマの増殖速度も明らかな抑制を認めた。
腫瘍組織から採取した腫瘍に浸潤している樹状細胞は、R848による刺激でIL-12を産生する能力を示した。
さらに、ジクロフェナクは腫瘍組織に浸潤している制御性T細胞(免疫細胞の活性を抑制する作用を示すT細胞)の数を減らし、制御性T細胞の活性化のマーカーであるCD25の発現を抑制した。
しかしながら、R848とジクロフェナクの腫瘍内への注入は、グリオーマ移植マウスの生存をさらに延長させる効果は認められなかった。
さらに検討すると、T細胞を活性化するときにジクロフェナクが存在するとT細胞からのINF-γの産生とT細胞の増殖を阻害した。これは、がんの免疫療法を行うとき、ジクロフェナクの投与の時期に注意する必要があることを示している。
以上をまとめると、ジクロフェナクはがん組織からの乳酸産生を抑制することによって、悪性神経膠腫における免疫抑制状態を改善する薬として興味深い作用を有している。
以下のような論文もあります。
New Aspects of an Old Drug – Diclofenac Targets MYC and Glucose Metabolism in Tumor Cells. (古い薬の新しい効果:ジクロフェナクはがん細胞のMYCとグルコース代謝をターゲットにする)PLoS One. 2013; 8(7): e66987.
【要旨】
ジクロフェナクのような非ステロイド性抗炎症剤は強力な抗がん作用を示す。現在まで、これらの作用は主にシクロオキシゲナーゼ(COX)の阻害作用との関連で議論されてきた。
この論文では、ジクロフェナクの新規のCOXに非依存的な作用を報告する。
ジクロフェナクはMYCの発現を有意に低下させ、グルコース代謝を制御し、培養細胞を使った実験ではメラノーマと白血病とがん細胞の細胞株で増殖を抑制し、in vivo(生体内)の実験でメラノーマの増殖を抑制した。
ジクロフェナクとは対照的に、非特異的なCOX阻害剤のアスピリンとCOX-2の選択的阻害剤のNS-398はMYC発現とグルコース代謝には何も影響しなかった。
ジクロフェナクはグルコーストランスポーター1(GLUT1)、乳酸脱水素酵素A(LDHA)、モノカルボン酸トランスポーター1(MCT1)の発現を有意に低下させ、その結果、グルコースの取込みと乳酸産生を低下させた。
これらの遺伝子発現の抑制に先行してジクロフェナクは乳酸の細胞内蓄積を亢進した。これは乳酸の排出に対してジクロフェナクが直接的な阻害作用を示すことを示唆している。
細胞内の乳酸の蓄積は細胞の増殖と遺伝子発現を抑制したが、MYCの発現は阻害しなかった。
テトラサイクリンでc-MYC遺伝子の発現を誘導する細胞を用いた実験で、ジクロフェナクはc-MYCの発現の有無にかかわらず、細胞増殖を抑制した。
このような結果から、ジクロフェナクはMYCと乳酸の輸送の阻害によって細胞の増殖を抑制することが示された。
これらの結果は、ジクロフェナクは臨床的に使用できるMYCと解糖系の阻害剤として既存の治療をサポートできる可能性を示している。
ジクロフェナク(Diclofenac)は商品名はボルタレンで、多数のジェネリック(後発品)もあって、安価な消炎鎮痛剤です。副作用に注意しなければなりませんが、がん細胞の解糖系をターゲットにしたがん治療で利用できるかもしれません。
LDH-AやMCTや解糖系をターゲットに、ジクロフェナクに低酸素誘導因子-1(HIF-1)の発現や活性を抑制するラパマイシンやジインドリルメタン、ピルビン酸脱水素酵素キナーゼを阻害してピルビン酸脱水素酵素を活性化するジクロロ酢酸ナトリウム、ヘキソキナーゼ活性を阻害する2-デオキシグルコース、呼吸酵素を阻害しミトコンドリアの酸化ストレスを高めるメトホルミンなどを併用するがん治療は理論的には可能性があると思います。 (乳酸脱水素酵素A(LDH-A)の阻害に関しては541話参照)
さて、この論文では、ジクロフェナクが「乳酸の排出に対してジクロフェナクが直接的な阻害作用を示す」ことが報告されています。
がん細胞から乳酸の排出を阻害し、がん細胞内の乳酸濃度が高まると乳酸脱水素酵素が阻害されるというメカニズムも想定されます。
がん細胞からの乳酸の排出を阻害する薬とメトホルミンを併用すると相乗的な抗がん作用があることが報告されています。最近、以下のような報告があり注目されています。
Dual Inhibition of the Lactate Transporters MCT1 and MCT4 Is Synthetic Lethal with Metformin due to NAD+ Depletion in Cancer Cells.(乳酸輸送体のMCT1とMCT4の二重阻害は、がん癌細胞におけるNAD +枯渇によって、メトホルミンとの併用で致死的に作用する)Cell Rep. 2018 Dec 11;25(11):3047-3058.
【要旨】
解糖系が高度に亢進したがん細胞は、モノカルボン酸トランスポーター1(MCT1)および4(MCT4)を介して解糖の最終産物である乳酸塩およびH+を排出することによって細胞内の酸性化を防止している。高血圧治療薬であるシロシンゴピン(syrosingopine,)は、MCT1とMCT4の両方を阻害する作用があり(MCT4に対して60倍高い阻害効力を持つ)、乳酸塩とH+の排出を阻害する。シロシンゴピンは、ミトコンドリアNADH脱水素酵素の阻害剤であるメトホルミンと相乗的な致死作用を示す。
解糖のATP生成段階に必要なNAD+は、ミトコンドリアNADH脱水素酵素または乳酸脱水素酵素によってNADHから再生される。
シロシンゴピン投与は、がん細胞内の乳酸濃度を高め、乳酸は乳酸脱水素酵素を阻害する。メトホルミンとシロシンゴピンの併用投与は、NAD+の再生を阻害し、解糖系での代謝を阻害し、ATP枯渇および細胞死を引き起こす。
NAD+やNAD前駆体のニコチンアミドモノヌクレオチド(NMN)やビタミンK2を外から供給するとATPレベルは部分的に回復できる。
したがって、メトホルミンと組み合わせたMCT1およびMCT4の薬理学的阻害は、潜在的ながん治療法となる。
メトホルミンは糖尿病の治療に最も多く使用されている薬です。血糖降下作用だけでなく、様々なメカニズムで抗がん作用を示すことが数多くの研究で明らかになっています。この論文の著者らは、2年前(2016年)に高血圧治療薬のシロシンゴピン(syrosingopine)がメトホルミンの抗がん作用を増強することを報告しています。
Syrosingopine sensitizes cancer cells to killing by metformin.(シロシンゴピンはがん細胞のメトホルミンによる細胞死誘導作用を増強する)
Sci Adv. 2016 Dec 23;2(12):e1601756.
その後の研究で、シロシンゴピンが乳酸トランスポーターによる乳酸排出を阻害して、がん細胞内の乳酸濃度を高め、その結果、乳酸脱水素酵素を阻害してNADHからNAD+の再生を阻害して、メトホルミンのがん細胞死誘導作用を増強するというメカニズムを明らかにしています。
つまり、メトホルミンとシロシンゴピンは異なるメカニズムでNAD+の量を減らす作用があり、相乗効果がある(がん細胞に致死的に働く組み合わせ:Lethal combination)という事です。
シロシンゴピンは1958年に降圧剤として開発されたようですが、現在では販売されていないので、入手はできません(ネットで検索した限りでは)。
この論文の著者らは、現時点では乳酸トランスポーターの阻害剤として使える薬が無いので、シロシンゴピンを乳酸トランスポーター阻害剤として開発するのかもしれません。
しかし、前述のジクロロフェナクは乳酸脱水素酵素を阻害してNAD+の再生を阻害するので、メトホルミンとジクロフェナクの併用は相乗的に抗がん作用を発揮すると思われます。
【メトホルミンはミトコンドリアの呼吸酵素複合体Iを阻害する】
糖尿病治療薬のメトホルミンはミトコンドリアの呼吸鎖の最初のステップである呼吸酵素複合体I を阻害することが明らかになっています。さらに、メトホルミンはミトコンドリアのグリセロールリン酸脱水素酵素(mitochondrial glycerol-phosphate dehydrogenase: mGDP)を阻害することも報告されています。その結果、ミトコンドリアでのATP産生を阻害します。NAD+(酸化型ニコチンアミド・アデニン・ジヌクレオチド)も低下します。
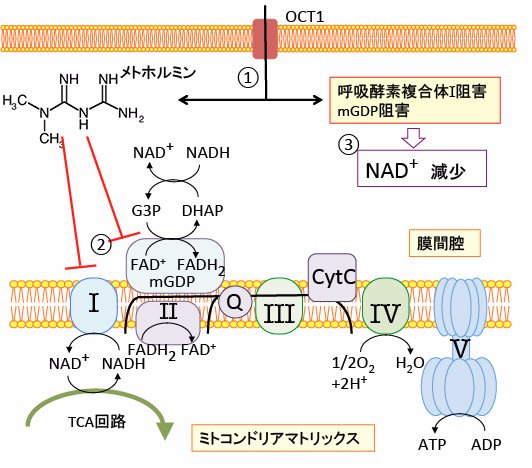
図:メトホルミンは有機カチオン輸送体1(organic cation transporter 1 :OCT1)によって細胞内に入り(①)、さらにミトコンドリアに集積する。ミトコンドリアでは呼吸酵素複合体IとmGDP(ミトコンドリア・グリセロールリン酸脱水素酵素)を阻害してNADHの酸化を阻害する(②)。その結果、ミトコンドリアでのATP産生は低下し、酸化型ニコチンアミド・アデニン・ジヌクレオチド(NAD+)が減少する(③)。
ATP産生が低下するとAMP:ATPの比が上昇し、AMP活性化プロテインキナーゼ(AMPK)を活性化します。活性化したAMPKは、肝臓の糖新生を抑制し、解糖を亢進し、骨格筋でのグルコース利用を促進して血糖を低下させます。
すなわち、メトホルミンの血糖降下作用はミトコンドリアにおけるATP産生の阻害によって体内のATP量が減少するためです。ATPを増やすために、グルコースの分解(異化)を促進し、糖新生(同化)を抑制するので血糖が低下します。
基本的には、運動と同じことです。運動も体内のATP量が減るので、グルコースの分解が促進されるように代謝が変わります。メトホルミンは運動しないで運動と同じ効果を発揮します。

図:AMP活性化プロテインキナーゼ(AMPK)は触媒作用を持つαサブユニットと、調節作用を持つβサブユットとγサブユニットから構成されるヘテロ三量体として存在する(①)。γサブユニットにはATPが結合しているが、ATPが枯渇してAMP/ATP比が上昇すると(②)、γサブユニットに結合していたATPがAMPに置き換わる(③)。その結果、アロステリック効果(酵素の立体構造が変化すること)によってこの複合体は中等度(2~10倍程度)に活性化され、AMPKキナーゼであるLKB1に対して親和性が高くなり、LKB1によってαサブユニットのスレオニン-172(Thr-172)がリン酸化され(④)、酵素活性は最大に活性化される。活性化したAMPKはインスリン感受性を亢進し、グルコースの取込みと分解を促進し、肝臓では糖新生を抑制して血糖を低下させる。さらに抗老化や寿命延長やがん予防の効果を発揮する(⑤)。運動やカロリー制限はATPを低下させAMP/ATP比を上昇してAMPKを活性化する(⑥)。メトホルミンはミトコンドリアの呼吸鎖を阻害してATP産生を低下させる機序とLKB1を活性化する両方の機序でAMPKを活性化する(⑦)。
メトホルミンは寿命を延ばす効果が知られています。この寿命延長効果は主にAMP活性化プロテインキナーゼ(AMPK)の活性化が関与しています。(384話参照)
AMPKはインスリンやインスリン様成長因子-1(IGF-1)によって活性が亢進するmTORC1(哺乳類ラパマイシン標的タンパク質複合体1)を抑制します。インスリン/IGF-1/TORC1シグナル伝達系は寿命を短くします。(362話参照)
つまり、運動やカロリー制限と同様にメトホルミンはAMPKを活性化する機序で、抗老化、寿命延長、がん予防の効果を発揮するのです。
【半枝蓮と高濃度ビタミンC点滴はPARPを活性化してはNADを低下する】
ポリ(ADP-リボース)ポリメラーゼ(PARP)は,核DNAに生じた一本鎖切断端を認識してDNAに結合して活性化され,ニコチンアミドアデニンジヌクレオチド(NAD+)を基質としてPARP自身やDNA修復関連タンパク質にADP-リボースを付加します(ポリ-ADP-リボシル化)。
ポリ-ADP-リボシル化はDNA修復反応を活性化しますが、過度のPARPの活性化はNAD+とATPの枯渇し細胞死(アポトーシス)を誘導します。
抗がん生薬の代表の半枝蓮はミトコンドリアでの活性酸素の産生を増やし、DNAの酸化障害からポリADPリボース合成酵素(PARP)が活性化され、NADを枯渇して解糖系を阻害してATPの産生を低下させ、酸化傷害によってミトコンドリアでの酸化的リン酸化によるATP産生も阻害するという機序が提案されています(下図)。

図:半枝蓮の成分はがん細胞のミトコンドリアでの活性酸素の産生を増やし(①)、酸化的リン酸化を阻害してATP産生を減らす(②)。さらに、DNAにダメージを与えてポリADPリボース合成酵素(PARP)を活性化し(③)、NADを枯渇して解糖系を阻害してATP産生を低下させる(④)。がん細胞のDNAダメージやATP枯渇によってがん細胞は死滅する(⑤)。
高濃度ビタミンC点滴は、1回に25〜100グラムという大量のビタミンCを1〜3時間かけて点滴する治療法です。がん細胞に取込まれたビタミンCが過酸化水素を発生することでDNAやミトコンドリアにダメージを与え、解糖系を阻害してATP産生を阻害することによって抗がん作用を発揮します。
ビタミンCはグルコースと構造が似ており、同じ糖輸送担体(グルコーストランスポーター)によって細胞内に取込まれます。がん細胞はグルコーストランスポーターの発現量が増え、グルコースの取込みが亢進しているので、大量のビタミンCががん細胞に取込まれ、がん細胞が選択的に死滅させることができます。
提唱されている作用機序として、ビタミンCによって発生した過酸化水素がDNAにダメージを与えると、ポリADPリボース合成酵素(PARP)が活性化されNADが枯渇し、解糖系もTCA回路も進まなくなります。活性酸素はミトコンドリアにもダメージを与えます。これらの作用で、エネルギーが枯渇して細胞が死滅することになります。前述の半枝蓮と似た機序です。
このようなメカニズムからOlaparibのようなポリADPリボース合成酵素(PARP)阻害剤は高濃度ビタミンC点滴の抗腫瘍効果を妨げる可能性があります。しかし、実際はPARP阻害剤と高濃度ビタミンC点滴は相乗効果が期待できるようです。以下のような報告があります。
Pharmacologic ascorbate induces neuroblastoma cell death by hydrogen peroxide mediated DNA damage and reduction in cancer cell glycolysis.(薬理学的アスコルビン酸は過酸化水素介在性のDNA損傷とがん細胞の解糖系の抑制によって神経芽細胞腫に細胞死を誘導する)Free Radic Biol Med. 2017 Dec;113:36-47.
【要旨】
アスコルビン酸は酸化ストレスを高め、腫瘍の増殖を遅らせることが示されている。この作用において、解糖系の抑制が起こることが推測されている。この研究では、この観察に関連するメカニズムをさらに検討した。
アスコルビン酸は過酸化水素を産生し、その結果、ATP枯渇とGAPDH(グリセルアルデヒド-3-リン酸デヒドロゲナーゼ)の阻害を引き起こして、ヒト神経芽細胞腫を死滅させる作用を示し、アポトーシスやオートファジーによる細胞死とは異なるタイプの細胞死を引き起こす。
細胞傷害性の機序は、PARP(ポリADPリボースポリメラーゼ)依存性DNA修復機構が活性化される場合と阻害された場合では異なっていた。
アスコルビン酸によって生成された過酸化水素はDNAを損傷し、PARPを活性化し、酸化型NAD(NAD+)を枯渇し、解糖系を阻害した。NAD+の補給は、ATP枯渇および細胞死を防止した。一方、PARP阻害剤のオラパリブ(olaparib)での処理は、NAD+およびATPレベルを維持したが、DNA二本鎖切断の増加をもたらし、アスコルビン酸誘発性細胞死を防止しなかった。
これらの実験結果は、正常なPARP関連DNA修復システムを有する細胞においては、アスコルビン酸誘導性細胞死はNAD+およびATP枯渇によって引き起こされるが、PARP活性が阻害された条件では、アスコルビン酸誘導性の細胞死は起こるが、それは活性酸素種によるDNA損傷の結果である。
マウス異種移植モデルでは、腹腔内に投与したアスコルビン酸は神経芽細胞腫の増殖を抑制し、生存期間を延長した。
以上をまとめると、これらのデータは、アスコルビン酸が解糖依存性腫瘍の治療に有効であり得ることを示唆している。また、解糖系以外の代替エネルギー代謝経路を使用するがんでは、PARP阻害剤をアスコルビン酸治療と組み合わせることが有用である。
高濃度のビタミンC(アスコルビン酸)は過酸化水素を発生してDNAにダメージを与え、PARPを活性化し、NAD+とATPを枯渇して細胞死を誘導します。そこで、PARPを阻害するとNADの枯渇が起きないので細胞死が起こらない可能性があります。
しかし、PARPの活性化が起こらない場合は、単純に活性酸素種によるDNA損傷で細胞死が誘導されるのです。OlaparibのようなPARP阻害剤を使用するとNAD+とATPの枯渇は起こらないが、DNA切断の修復ができないので、細胞死が誘導されるということです。
つまり、PARP阻害と高濃度ビタミンC点滴の併用は、抗がん作用を強化できるということです。
血中の濃度がmMレベルに上昇する高濃度のビタミンCの点滴は、ビタミンとしての働きではなく、抗がん剤としての働きを発揮します。この薬理学的濃度のビタミンCは過酸化水素を発生し、フェントン反応によってヒドロキシルラジカルのような酸化傷害を引き起こす活性酸素種を産生してDNAを損傷するためです。この作用機序を下図にまとめています。

図:ビタミンCはグルコーストランスポーターから細胞内に取込まれる。がん細胞はグルコーストランスポーターの発現量が増えているので、がん細胞に高用量のビタミンCが取込まれる(①)。取込まれたビタミンCはがん細胞内で過酸化水素(H2O2)を発生させて、DNAとミトコンドリアにダメージを与える(②)。DNAのダメージはポリADPリボース合成酵素(PARP)の活性を亢進してNAD+(ニコチンアミドアデニンジヌクレオチド)が減少し(③)、解糖系が阻害される(④)。ミトコンドリアのダメージは酸化的リン酸化でのATP産生を減少させる(⑤)。この結果、がん細胞内のATPが枯渇してがん細胞は死滅する(⑥)。PARP阻害剤を使用するとNAD+の枯渇は起こらないが、DNA修復ができないので、細胞死が誘導される(⑦)。したがって、PARP阻害剤と高濃度ビタミンC点滴の併用は相乗効果が期待できる。
高濃度のビタミンCががん細胞にダメージを与えて、正常細胞にはダメージを与えないのは、がん細胞が解糖系が亢進していることが主な理由になっています。つまり、がん細胞は解糖系でのATP産生に依存度が高いためです。
高濃度のビタミンCは過酸化水素を産生して、活性酸素の産生を増やしてDNAを損傷します。DNAを損傷するとPARPが活性化されて、NAD+が消耗されます。NAD+が消耗して枯渇すると解糖系での代謝が進まなくなりATPも枯渇します。
PARP阻害剤はNAD+とATPの枯渇を阻止しますが、致死的なDNA損傷によって細胞死を引きこします。
以上をまとめると、高濃度ビタミンC点滴を行なっているときにメトホルミン、ジクロフェナク、半枝蓮などの併用は、NAD+とATPの枯渇作用を増強して、相乗効果が期待できます。PARP阻害剤のOlaparibとの併用も問題ありません。
| « 632)漢方治療... | 634)シンバス... » |