E. Ntzani, J. Ioannidis
Predictive ability of DNA microarrays for cancer outcomes and correlates: an empirical assessment.
The Lancet, Volume 362, Issue 9394, Pages 1439-1444, 2003
[PDF]
・ガンの診断に関するマイクロアレイデータを網羅的に集め、その解析方法やデータの大きさなどの実験環境と診断の精度との関係について検証する。
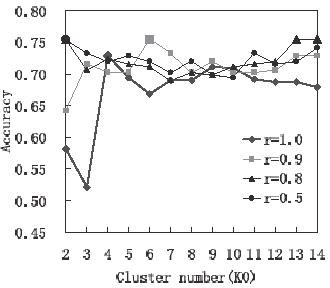
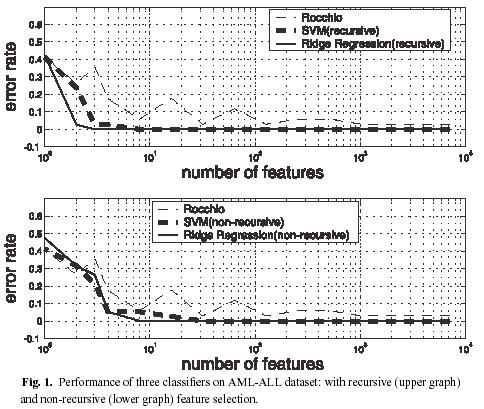
・データ:84種類。
・結論「DNA microarrays addressing cancer outcomes show variable prognostic performance. Larger studies with appropriate clinical design, adjustment for known predictors, and proper validation are essential for this highly promising technology.」
Predictive ability of DNA microarrays for cancer outcomes and correlates: an empirical assessment.
The Lancet, Volume 362, Issue 9394, Pages 1439-1444, 2003
[PDF]
・ガンの診断に関するマイクロアレイデータを網羅的に集め、その解析方法やデータの大きさなどの実験環境と診断の精度との関係について検証する。
・データ:84種類。
・結論「DNA microarrays addressing cancer outcomes show variable prognostic performance. Larger studies with appropriate clinical design, adjustment for known predictors, and proper validation are essential for this highly promising technology.」