Satoshi Niijima and Satoru Kuhara
Recursive gene selection based on maximum margin criterion: a comparison with SVM-RFE
BMC Bioinformatics 2006, 7:543
[PDF][Web Site]
・マイクロアレイデータによるサンプル識別法として、Maximum margin criterion (MMC)の判別ベクトルを利用した再帰的遺伝子抽出法(RFE; recursive feature elimination)を提案する。これは古典的な線形判別分析の応用である。
・データ
(Binary-class)
1.Colon cancer [Alon]
2.Prostate cancer [Singh]
3.Leukemia [Golub]
4.Medulloblastoma [Pomeroy]
5.Breast cancer [van'tVeer]
(Multi-class)
6.MLL [Armstrong]
7.SRBCT [Khan]
8.CNS [Pomeroy]
9.NCI60 [Ross]
・比較法:SVM-RFE
・識別結果の評価法:3分割 cross-validation、100回繰り返し
・動機「Gene selection plays essential roles in classification tasks. It improves the prediction accuracy of classifiers by using only discriminative genes. It also saves computational costs by reducing dimensionality. More importantly, if it is possible to identify a small subset of biologically relevant genes, it may provide insights into understanding the underlying mechanism of a specific biological phenomenon. Also, such information can be useful for designing less expensive experiments by targeting only a handful of genes.」
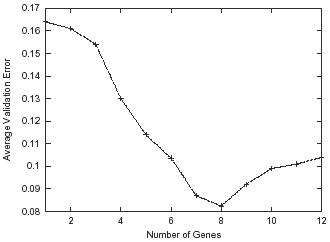
・注意「In this study, we do not address the problem of finding the optimum number of genes that would yield highest classification accuracy. Instead, the number of genes was varied from 1 to 100, and the performances were compared for each number of genes.」
・結果「As our results indicate, the prediction of clinical outcome is generally more difficult than that of tissue or disease types.」
・結果「The results suggest that MMC-RFE is less sensitive to noise and outliers due to the use of average margin, while the performance of SVM-RFE can be easily affected by them when applied to noisy, small sample size microarray data. Another advantage of MMC-RFE over SVM-RFE is that MMC-RFE naturally extends to multi-class cases. Furthermore, MMC-RFE does not require the computation of the matrix inversion unlike LDA-RFE and MSE-RFE, and involves no parameters to be tuned.」
・RFEとは「The idea of recursive feature elimination (RFE) [6] is to recursively remove genes using the absolute weights of the discriminant vector or hyperplane, which reflect the significance of the genes for classification.」
Recursive gene selection based on maximum margin criterion: a comparison with SVM-RFE
BMC Bioinformatics 2006, 7:543
[PDF][Web Site]
・マイクロアレイデータによるサンプル識別法として、Maximum margin criterion (MMC)の判別ベクトルを利用した再帰的遺伝子抽出法(RFE; recursive feature elimination)を提案する。これは古典的な線形判別分析の応用である。
・データ
(Binary-class)
1.Colon cancer [Alon]
2.Prostate cancer [Singh]
3.Leukemia [Golub]
4.Medulloblastoma [Pomeroy]
5.Breast cancer [van'tVeer]
(Multi-class)
6.MLL [Armstrong]
7.SRBCT [Khan]
8.CNS [Pomeroy]
9.NCI60 [Ross]
・比較法:SVM-RFE
・識別結果の評価法:3分割 cross-validation、100回繰り返し
・動機「Gene selection plays essential roles in classification tasks. It improves the prediction accuracy of classifiers by using only discriminative genes. It also saves computational costs by reducing dimensionality. More importantly, if it is possible to identify a small subset of biologically relevant genes, it may provide insights into understanding the underlying mechanism of a specific biological phenomenon. Also, such information can be useful for designing less expensive experiments by targeting only a handful of genes.」
・注意「In this study, we do not address the problem of finding the optimum number of genes that would yield highest classification accuracy. Instead, the number of genes was varied from 1 to 100, and the performances were compared for each number of genes.」
・結果「As our results indicate, the prediction of clinical outcome is generally more difficult than that of tissue or disease types.」
・結果「The results suggest that MMC-RFE is less sensitive to noise and outliers due to the use of average margin, while the performance of SVM-RFE can be easily affected by them when applied to noisy, small sample size microarray data. Another advantage of MMC-RFE over SVM-RFE is that MMC-RFE naturally extends to multi-class cases. Furthermore, MMC-RFE does not require the computation of the matrix inversion unlike LDA-RFE and MSE-RFE, and involves no parameters to be tuned.」
・RFEとは「The idea of recursive feature elimination (RFE) [6] is to recursively remove genes using the absolute weights of the discriminant vector or hyperplane, which reflect the significance of the genes for classification.」