Alan R. Dabney
Classification of microarrays to nearest centroids
Bioinformatics 2005 21(22):4148-4154
[PDF][Web Site]
・サンプルのクラス分け法として、Classification to Nearest Centroids (ClaNC)を提案する。LDAに基づくアルゴリズムの簡便さが特長。
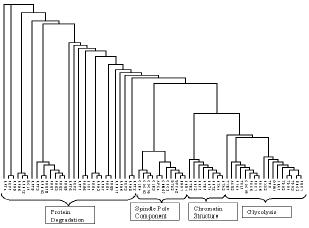
・人工データ:4クラス、各クラス30サンプル、5000遺伝子、発現量の分布を変化させた3種のデータを用意
・生データ
1.Small round blue cell tumors (SRBCT)、2307遺伝子、83サンプル、4クラス [Khan]
2.Lymphoma、4026遺伝子、58サンプル、3クラス [Alizadeh]
3.NCI cancer cell lines、6830遺伝子、60サンプル、10クラス [Ross]
4.Leukemia、3857遺伝子、38サンプル、2クラス [Golub]
・クラス分けの比較法:Prediction Analysis of Microarrays (PAM)[Tibshirani]、デフォルト使用の他に設定を変えた4つの方法を使用
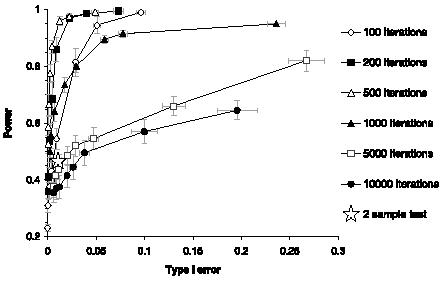
・クラス分けの評価法(error rates):5-fold cross-validation
・問題点「I surprisingly show that the modified t-statistics and shrunken centroids employed by PAM tend to increase misclassification error when compared with their simpler counterparts.」
・問題点「For example, with unlimited resources, we may wish to use all relevant genes in the classifier; although, we may be able to find a subset of genes that classify just as well as (or even better than) the complete set. In other settings, it may be necessary to make tradeoffs between accuracy and practicality.」
・方法「I present here an alternative LDA-based classifier that I call ClaNC, for Classification to Nearest Centroids. ClaNC (1) does not shrink centroids, (2) uses unmodified t-statistics to select genes, (3) carries out class-specific feature selection, and (4) allows each gene to be active in at most one class.」
・結果「LDA-based classifiers that are even simpler than PAM can perform very well.」
・展望「I intend to perform a more thorough investigation of shrinkage for classification in future work.」
Classification of microarrays to nearest centroids
Bioinformatics 2005 21(22):4148-4154
[PDF][Web Site]
・サンプルのクラス分け法として、Classification to Nearest Centroids (ClaNC)を提案する。LDAに基づくアルゴリズムの簡便さが特長。
・人工データ:4クラス、各クラス30サンプル、5000遺伝子、発現量の分布を変化させた3種のデータを用意
・生データ
1.Small round blue cell tumors (SRBCT)、2307遺伝子、83サンプル、4クラス [Khan]
2.Lymphoma、4026遺伝子、58サンプル、3クラス [Alizadeh]
3.NCI cancer cell lines、6830遺伝子、60サンプル、10クラス [Ross]
4.Leukemia、3857遺伝子、38サンプル、2クラス [Golub]
・クラス分けの比較法:Prediction Analysis of Microarrays (PAM)[Tibshirani]、デフォルト使用の他に設定を変えた4つの方法を使用
・クラス分けの評価法(error rates):5-fold cross-validation
・問題点「I surprisingly show that the modified t-statistics and shrunken centroids employed by PAM tend to increase misclassification error when compared with their simpler counterparts.」
・問題点「For example, with unlimited resources, we may wish to use all relevant genes in the classifier; although, we may be able to find a subset of genes that classify just as well as (or even better than) the complete set. In other settings, it may be necessary to make tradeoffs between accuracy and practicality.」
・方法「I present here an alternative LDA-based classifier that I call ClaNC, for Classification to Nearest Centroids. ClaNC (1) does not shrink centroids, (2) uses unmodified t-statistics to select genes, (3) carries out class-specific feature selection, and (4) allows each gene to be active in at most one class.」
・結果「LDA-based classifiers that are even simpler than PAM can perform very well.」
・展望「I intend to perform a more thorough investigation of shrinkage for classification in future work.」