








Trond Hellem B? and Inge Jonassen
New feature subset selection procedures for classification of expression profiles
Genome Biology 2002, 3:research0017.1-0017.11
[PDF][Web Site]
・サンプルのクラス分けに使う遺伝子の抽出法の提案。サンプルを最もよく分離できる二つ一組(pair)の遺伝子を抽出(→繰り返す)。
・データ
1.Leukemia, 72 samples (ALL 47/ALL 25), 7129 genes [Golub]
2.Colon cancer, 62 samples (normal 22/tumor 40), 6500 genes [Alon]
・遺伝子ランキング法
1.Individual ranking (IR, 従来法)
2.Forward selection (FS, 従来法)
3.All pairs (AP, 提案法):全ての遺伝子について総あたりで計算
4.Greedy pairs (GP, 提案法):t-scoreでふるいにかけた遺伝子のみで計算、処理を高速化
・識別法(サンプルクラス分け)
1.Diagonal linear discriminant (DLD)
2.Fisher's linear discriminant (FLD)
3.k nearest neighbors (kNN)
・ソフトウェア:J-Express software packageで利用可能
・概要「Our results have implications for how to select marker genes and how many gene measurements are needed for diagnostic purposes.」
・問題点「One problem with gene expression data is that each example has too many features, and many of them are noisy and irrelevant for the learning problem at hand.」
・FSSとは「The problem of finding the best subset is commonly referred to as the feature subset selection (FSS) problem.」
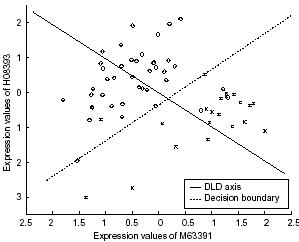
・方法「We give each pair of genes a score reflecting how well the pair in combination distinguishes two experiment classes.」
・処理「We evaluate a gene pair by computing the projected coordinates of each experiment on the DLD axis using only these two genes. We then take the two sample t-statistic on the projected points as the pair socre.」
・問題点「We do not claim that our pair-based methods will find all interesting genes, as there may be relevant genes that are significant by themselves but may not appear in any of the high-scoring pairs.」
・前処理「Before analysis, we carried out the following preprocessing steps on both datasets: base 10 logarithmic trasformation; and for each gene, subtract the mean and divide by the standard deviation.」
・遺伝子ランキング法のまとめ「A large number of measures have been proposed for scoring genes, starting with Golub et al.[2] that proposed using |(μ1-μ2)/(σ1+σ2)|. Other gene measures in the literature include both non-parametric measures like the TNoM score of Ben-Dor et al.[17] and information gain (proposed by Xing et al. [6]), and parametric measures like t-score[7], Fisher score[13], naive Bayes global relevance [18] and between- to within-variance ratio [16].」
・この場合も識別率が悪い所から始まって指数関数的に上昇する。なぜ?
New feature subset selection procedures for classification of expression profiles
Genome Biology 2002, 3:research0017.1-0017.11
[PDF][Web Site]
・サンプルのクラス分けに使う遺伝子の抽出法の提案。サンプルを最もよく分離できる二つ一組(pair)の遺伝子を抽出(→繰り返す)。
・データ
1.Leukemia, 72 samples (ALL 47/ALL 25), 7129 genes [Golub]
2.Colon cancer, 62 samples (normal 22/tumor 40), 6500 genes [Alon]
・遺伝子ランキング法
1.Individual ranking (IR, 従来法)
2.Forward selection (FS, 従来法)
3.All pairs (AP, 提案法):全ての遺伝子について総あたりで計算
4.Greedy pairs (GP, 提案法):t-scoreでふるいにかけた遺伝子のみで計算、処理を高速化
・識別法(サンプルクラス分け)
1.Diagonal linear discriminant (DLD)
2.Fisher's linear discriminant (FLD)
3.k nearest neighbors (kNN)
・ソフトウェア:J-Express software packageで利用可能
・概要「Our results have implications for how to select marker genes and how many gene measurements are needed for diagnostic purposes.」
・問題点「One problem with gene expression data is that each example has too many features, and many of them are noisy and irrelevant for the learning problem at hand.」
・FSSとは「The problem of finding the best subset is commonly referred to as the feature subset selection (FSS) problem.」
・方法「We give each pair of genes a score reflecting how well the pair in combination distinguishes two experiment classes.」
・処理「We evaluate a gene pair by computing the projected coordinates of each experiment on the DLD axis using only these two genes. We then take the two sample t-statistic on the projected points as the pair socre.」
・問題点「We do not claim that our pair-based methods will find all interesting genes, as there may be relevant genes that are significant by themselves but may not appear in any of the high-scoring pairs.」
・前処理「Before analysis, we carried out the following preprocessing steps on both datasets: base 10 logarithmic trasformation; and for each gene, subtract the mean and divide by the standard deviation.」
・遺伝子ランキング法のまとめ「A large number of measures have been proposed for scoring genes, starting with Golub et al.[2] that proposed using |(μ1-μ2)/(σ1+σ2)|. Other gene measures in the literature include both non-parametric measures like the TNoM score of Ben-Dor et al.[17] and information gain (proposed by Xing et al. [6]), and parametric measures like t-score[7], Fisher score[13], naive Bayes global relevance [18] and between- to within-variance ratio [16].」
・この場合も識別率が悪い所から始まって指数関数的に上昇する。なぜ?




